
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Warum Nanopore-Amplikon-Sequenzierung wichtig ist
Kurzleseverfahren, wie Illumina-Panels, die auf die V3–V4-Regionen abzielen, können oft nicht zwischen eng verwandten Arten unterscheiden. Dies kann die nachgelagerte Forschung in der Mikrobiomanalyse, der Pathogenerkennung und der klonalen Validierung einschränken.
Nanopore-Sequenzierung schließt diese Lücken, indem sie produziert lange Lesungen über die gesamte Genlängewie V1–V9 des 16S oder die gesamte ITS-Region. Diese Reads bieten eine höhere taxonomische Auflösung, reduzieren Fehler in der Gemeinschaftsanalyse und ermöglichen eine zuverlässige Identifizierung auf Arten- oder Stammniveau.
Für Forscher, die einen breiteren Kontext benötigen, Nanopore Ultra-Lang Sequenzierung unterstützt Telomer-zu-Telomer-Genomassemblierungen, während Nanopore-Direkt-RNA-Sequenzierung Linksstruktur mit Ausdruck.
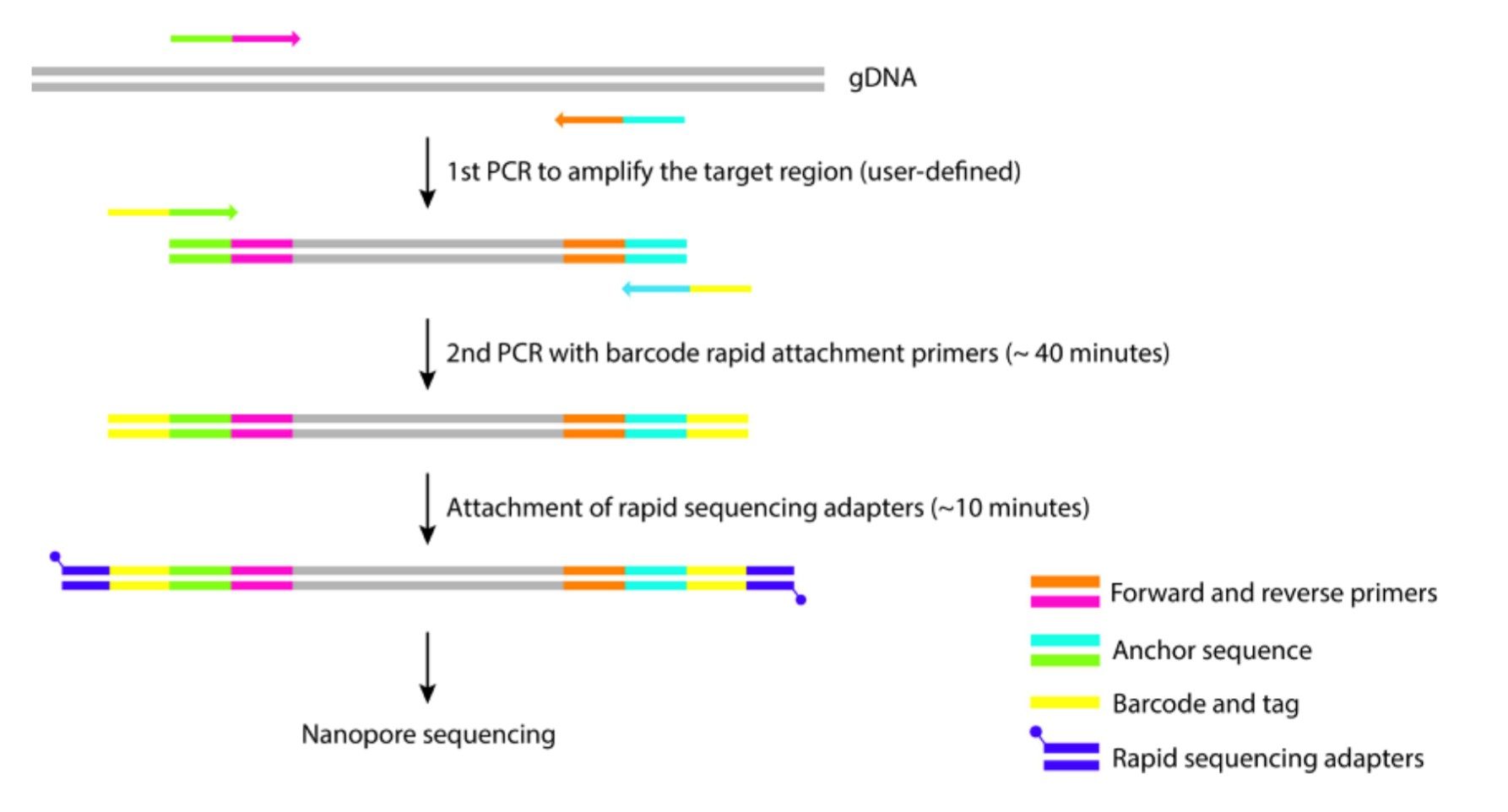 Workflow für den zweistufigen PCR-Ansatz in der Nanopore-Amplikon-Sequenzierungsbibliotheksvorbereitung. (Diese Abbildung wurde von Yoshiyuki Matsuo (https://dx.doi.org/10.17504/protocols.io.8epv5zrodv1b/v4) angepasst.)
Workflow für den zweistufigen PCR-Ansatz in der Nanopore-Amplikon-Sequenzierungsbibliotheksvorbereitung. (Diese Abbildung wurde von Yoshiyuki Matsuo (https://dx.doi.org/10.17504/protocols.io.8epv5zrodv1b/v4) angepasst.)
Technische Spezifikationen
CD Genomics bietet einen zuverlässigen, durchgängigen Workflow für Nanopore-Amplikon-Sequenzierung, optimiert für sowohl klonale als auch gemeinschaftsbasierte Projekte. Unsere Laborprotokolle, Sequenzierungsplattformen und bioinformatischen Pipelines sind darauf ausgelegt, die Lesegenauigkeit zu maximieren und eine Auflösung auf Arten- oder Stammniveau zu gewährleisten.
| Parameter | Spezifikation |
|---|---|
| Amplicon-Größenbereich | 0,5–25 kb (erweiterte Unterstützung auf Anfrage verfügbar) |
| Plattformen | Oxford Nanopore PromethION / GridION |
| Chemie | Kit 14 mit R10.4.1 Flusszellen für verbesserte Genauigkeit |
| Lese-Tiefe | ≥50.000 Reads pro Probe empfohlen für die Gemeinschaftsprofilierung |
| Durchlaufzeit | 1–3 Werktage (klonale Projekte); ~14 Tage (Gemeinschaftsprofilierung) |
| Datengenauigkeit | Standardpolierpipelines; optional barcode-basiertes Konsens-Workflow für höhere Genauigkeit |
| Liefergegenstände | FASTQ/FASTA-Dateien, Qualitätskontrollberichte, Taxonomie-/Varianten-Tabellen, interaktive HTML- und PDF-Berichte |
QualitätssicherungAlle Sequenzierungsprojekte unterliegen strengen Qualitätskontrollen, einschließlich DNA-Integritätsprüfungen, Analyse der Lese-Längenverteilung und Überwachung der Fehlerquote.
Anwendungen
Nanopore-Amplikon-Sequenzierung wird häufig angewendet in mikrobiologische ForschungGenvalidierung und vergleichende GenomikDurch die Bereitstellung von langen Reads, die gesamte Amplicons abdecken, ermöglicht es eine zuverlässige Interpretation komplexer Datensätze und verringert die Mehrdeutigkeit, die häufig bei Kurzleseverfahren auftritt.
Mikrobielle Gemeinschaftsprofilierung
- Vollständige 16S rRNA (V1–V9) und ITS-Regionen liefern eine Identifizierung auf Artenebene.
- Geeignet für Umweltproben, menschliche Mikrobiome und industrielle Mikrobiologieprojekte.
- Unterstützt die Diversitätsanalyse, die Bewertung der Populationsstruktur und vergleichende Studien.
Pathogenüberwachung und Risikobewertung
- Erkennen Sie potenzielle Krankheitserreger in Wasser, Boden, Lebensmitteln und Innenräumen.
- Ermöglicht die Verfolgung von mikrobiellen Veränderungen als Reaktion auf saisonale oder umweltbedingte Veränderungen.
- Verbessert die Biosicherheitsüberwachung in der klinischen und landwirtschaftlichen Forschung.
Lebensmittelsicherheit und Landwirtschaft
- Identifizieren Sie Verderborganismen und nützliche Stämme in Lebensmittelproduktionsketten.
- Unterstützen Sie Zuchtprogramme, indem Sie mikrobielle Gemeinschaften profilieren, die mit der Resilienz von Pflanzen verbunden sind.
- Erleichtern Sie Studien zur landwirtschaftlichen Biotechnologie, indem Sie die Diversität auf Stamm-Ebene auflösen.
Klonale Verifizierung und Variantenentdeckung
- Bestätigen Sie Einzelgen-Amplicons, bearbeitete Konstrukte oder Plasmid-Einfügungen.
- Generieren Sie hochpräzisen Konsens für klonale Proben mithilfe von barcode-basierten Strategien.
- Schnelle Durchlaufzeiten ermöglichen iterative Design- und Testzyklen.
Vergleichende und evolutionäre Genomik
- Lösen Sie eng verwandte Arten und Varianten auf Stammebene.
- Unterstützen Sie Studien zur Populationsgenetik und mikrobiellen Evolution mit vollständiger Abdeckung.
- Kombinieren mit Nanopore-Zielsequenzierung für eine gezielte Analyse spezifischer Loci.
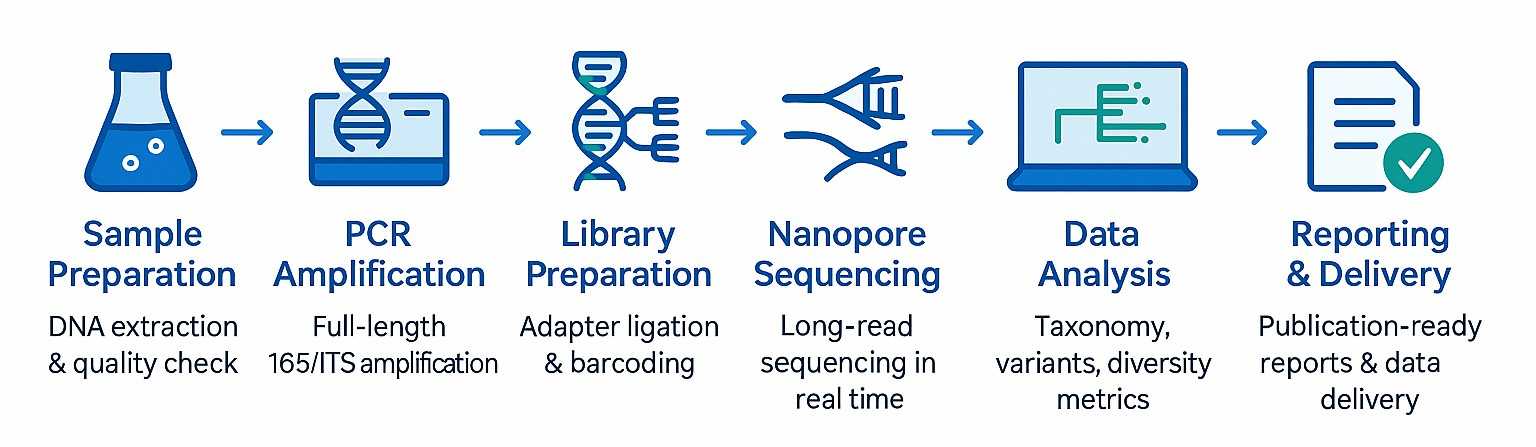
Workflow: End-to-End-Service
Primer-Strategie: Zielregionen auswählen (16S, ITS, benutzerdefinierte Gene)
Hochpräzise PCR: mit optionaler Barcode-Kennzeichnung zur Fehlerkorrektur
BibliotheksvorbereitungONT-Kit 14, minimale Fragmentierung
SequenzierungPromethION/GridION mit R10.4.1 Flusszellen
DatenverarbeitungBasisaufruf, Konsensgenerierung, Chimärenfilterung
Bioinformatiktaxonomische Zuordnung, Variantenanalyse, Diversitätsstatistiken
LieferungFASTQ/FASTA-Dateien, QC-Berichte, HTML/PDF-Ausgaben
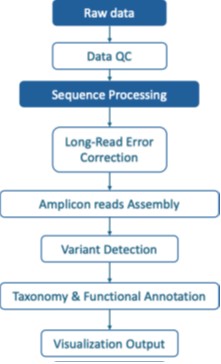
Bioinformatik & Berichterstattung
CD Genomics bietet eine vollständige Bioinformatik-Pipeline für Nanopore-Amplikon-Sequenzierung, um Ergebnisse mit hoher Zuverlässigkeit und veröffentlichungsfähige Ausgaben zu gewährleisten. Unsere Analyse umfasst Qualitätskontrolle, taxonomische Klassifikation, Variantenerkennung und maßgeschneiderte Berichterstattung.
Standardanalyse
Basisaufruf und DemultiplexingUmwandlung von Rohsignalen in FASTQ mit Proben-Trennung.
Qualitätskontrolle: Verteilung der Lese-Längen, Abdeckungs-Tiefe und Genauigkeitsstatistiken.
Taxonomische ZuordnungArtenklassifikation für 16S- und ITS-Datensätze unter Verwendung kuratierter Referenzdatenbanken.
DiversitätsanalyseAlpha- und Beta-Diversitätsindizes mit Visualisierungen wie PCoA- oder NMDS-Diagrammen.
Erweiterte Analyse (Optional)
Differenzielle HäufigkeitVergleichende Statistiken zwischen den experimentellen Gruppen.
VariantenerkennungIdentifizieren von Sequenzvarianten innerhalb klonaler Amplicons.
Barcode-Konsens-Workflowverbesserte Genauigkeit durch Fehlerunterdrückung und Chimärenfilterung.
Stammauflösungsniveau: Clustering und feinkalibrige Variantenbestimmung.
Bewährte Anwendungsfälle
Unser Nanopore-Amplikon-Sequenzierung Der Service wurde erfolgreich in verschiedenen Forschungsbereichen angewendet. Im Folgenden sind ausgewählte Beispiele aufgeführt, die praktische Ergebnisse zeigen.
| Projekt | Forschungsziel | Schlüsselergebnisse |
|---|---|---|
| Städtisches Wasser-Mikrobiom | Bewerten Sie saisonale Veränderungen und die Auswirkungen von Verschmutzung auf Flussgemeinschaften. | Artenebene Auflösung von Arcobacter und Aeromonas; zeigte quellenspezifische Beiträge in nassen vs. trockenen Jahreszeiten |
| Studie zur Innenraumumgebung | Charakterisierung mikrobieller Populationen, die mit menschlicher Besiedlung verbunden sind. | 21 Schlüsselarten identifiziert, darunter 11 potenzielle Krankheitserreger; höhere Häufigkeit von nützlichen Bakterien in Innenräumen. |
| Klonale Genverifizierung | Bestätigen Sie konstruierte Konstrukte und Plasmidsequenzen. | Hochpräziser Konsens erreicht; reduzierte Fehlalarme durch barcodebasierte Strategie |
| Agrar-Mikrobiom | Untersuchen Sie die mit Pflanzen verbundenen mikrobiellen Diversität in Bodenproben. | Verbesserte Stämmebene-Profilierung; unterstützte Resilienzanalysen in der Züchtungsforschung |
HinweisDiese Anwendungen sind für Nur für Forschungszwecke und sind nicht für diagnostische Verfahren bestimmt.
Liefergegenstände
- Rohsequenzierungsdaten: FASTQ- und optionale FASTA-Dateien.
- QC-Berichte: Ausbeute, Verteilung der Lese-längen und Genauigkeitsmetriken.
- Taxonomie/Variantentabellen mit Abundanzprofilen.
- Interaktive HTML- und PDF-Berichte mit veröffentlichungsfertigen Abbildungen (SVG/PNG).
- Methoden-Text und Pipeline-Parameter zur Unterstützung der Reproduzierbarkeit.
Die Wahl der richtigen Sequenzierungsplattform
CD Genomics unterstützt mehrere Technologien für Lang- und Kurzlesungen und stellt sicher, dass jedes Projekt den geeignetsten Ansatz erhält. Egal, ob Ihr Ziel die hochgenaue Variantenentdeckung, die Analyse von Voll-Längen-Ampliconen oder Studien im Bevölkerungsausmaß ist, wir können die richtige Plattform empfehlen.
| Merkmal | PacBio SMRT | Oxford Nanopore | Illumina |
|---|---|---|---|
| Leseumfang | Lang (10–25 kb; HiFi ~15–20 kb) | Ultra-lang (kb bis Mb-Bereich) | Kurz (100–500 bp) |
| Genauigkeit | Sehr hoch (HiFi >99,9%) | Hoch mit Konsens-/Fehlerunterdrückung | Sehr hoch (>99,9%) |
| Wende | Moderat | Flexibel, tragbar, in Echtzeit | Hochdurchsatz, batchbasiert |
| Stärken | Geringe Fehlerquote, ideal für sich wiederholende Regionen | Längste Reads, Artenebene Amplicons, direkte RNA/DNA | Kostenwirksam für große Kohorten |
| Einschränkungen | Höhere Kosten, DNA-Qualität entscheidend | Die rohe Fehlerquote ist ohne Korrektur höher. | Begrenzte Auflösung für lange Wiederholungen |
| Am besten geeignet für | De-novo-Assemblierungen, Wiederholungsregionen | Amplicons, Mikrobiome, schnelle Analyse | SNP-Erkennung, große Stichprobenstudien |
Unser Engagement:
Wir bieten Sequenzierungsdienste an. alle drei Plattformen—PacBio, Oxford Nanopore und Illumina—daher müssen Sie sich keine Sorgen machen, die falsche Methode auszuwählen. Unsere Experten bewerten Ihre Proben und Forschungsziele und empfehlen dann die Plattform, die die Datenqualität, Effizienz und Kosteneffektivität maximiert.
Beispielanforderungen für Nanopore-Amplikon-Sequenzierung
| Probenart | Empfohlene Eingabe | Reinheit (OD260/280) | Anforderungen | Notizen |
| Reinige PCR-Produkte | ≥1 μg (min. 500 ng) | 1,8–2,0 | ≥20 ng/μL; hochqualitativ; Größe entspricht den Plattform-Spezifikationen | Einzelne, spezifische Band; keine unspezifischen Produkte |
| Unreinigte PCR-Produkte | Variable | — | Akzeptabel, aber eine Reinigung wird dringend empfohlen, um eine optimale Sequenzierungsqualität zu gewährleisten. | — |
| Fragmentierte DNA | Ausreichende Menge | — | Erfordert einheitliche Größe und Kompatibilität mit der Zielregion. | Für spezifische Amplicon-Strategien |
| Genomische DNA (gDNA) | ≥500 ng | 1,8–2,0 | Hohe Reinheit; ≥20 ng/μL; keine Zersetzung | Ideal für PCR-basierte Amplifikation |
- Alle DNA-Proben müssen auf Reinheit und Konzentration getestet werden, um die Sequenzierungsqualität sicherzustellen.
- Wenn Sie Fragen zur Probenvorbereitung haben oder einen maßgeschneiderten Plan benötigen, zögern Sie nicht, uns jederzeit für fachkundige Unterstützung zu kontaktieren.
Warum CD Genomics für Nanopore-Amplikon-Sequenzierung wählen?
Von fortschrittlichen Sequenzierungsplattformen bis hin zu hochwertigen Datenlieferungen bietet CD Genomics eine effiziente, end-to-end Lösung, die auf vielfältige Forschungsbedürfnisse zugeschnitten ist. Egal, ob Sie seltene Varianten untersuchen oder alte DNA sequenzieren, unser Team sorgt für zuverlässige Ergebnisse mit flexibler Unterstützung.
- ExpertiseUnser Team verfügt über umfassende Expertise in der Nutzung der Technologie von Oxford Nanopore für verschiedene Sequenzierungsanwendungen.
- SpitzentechnologieWir nutzen die neuesten Fortschritte in der Nanoporen-Sequenzierung, um die bestmöglichen Ergebnisse zu liefern.
- Anpassbare Lösungen: Wir passen jedes Projekt an die spezifischen Bedürfnisse unserer Kunden an und gewährleisten relevante und qualitativ hochwertige Ergebnisse.
- Schnelle BearbeitungszeitMit Echtzeit-Sequenzierungsfähigkeiten bieten wir schnelle Einblicke, um Ihre Forschung auf Kurs zu halten.
- Umfassende UnterstützungVon der Probenvorbereitung bis zur Datenanalyse bieten wir umfassende Unterstützung während des gesamten Sequenzierungsprozesses.
Demo-Ergebnisse
Demonstrationsergebnisse Präsentation
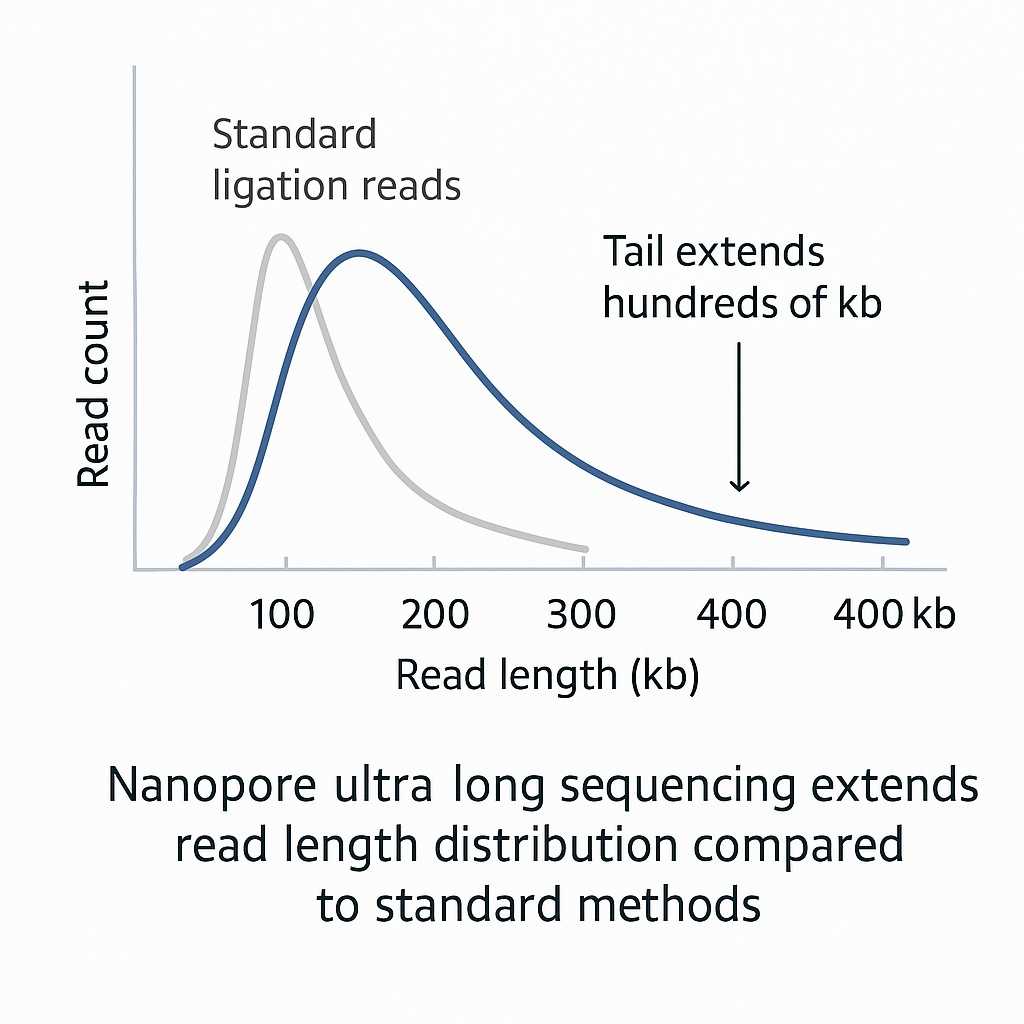
Nanopore-Lesungen umfassen die gesamten 16S/ITS-Regionen, während Kurzleseplattformen auf partielle Segmente beschränkt sind.
Verbesserung der taxonomischen Auflösung
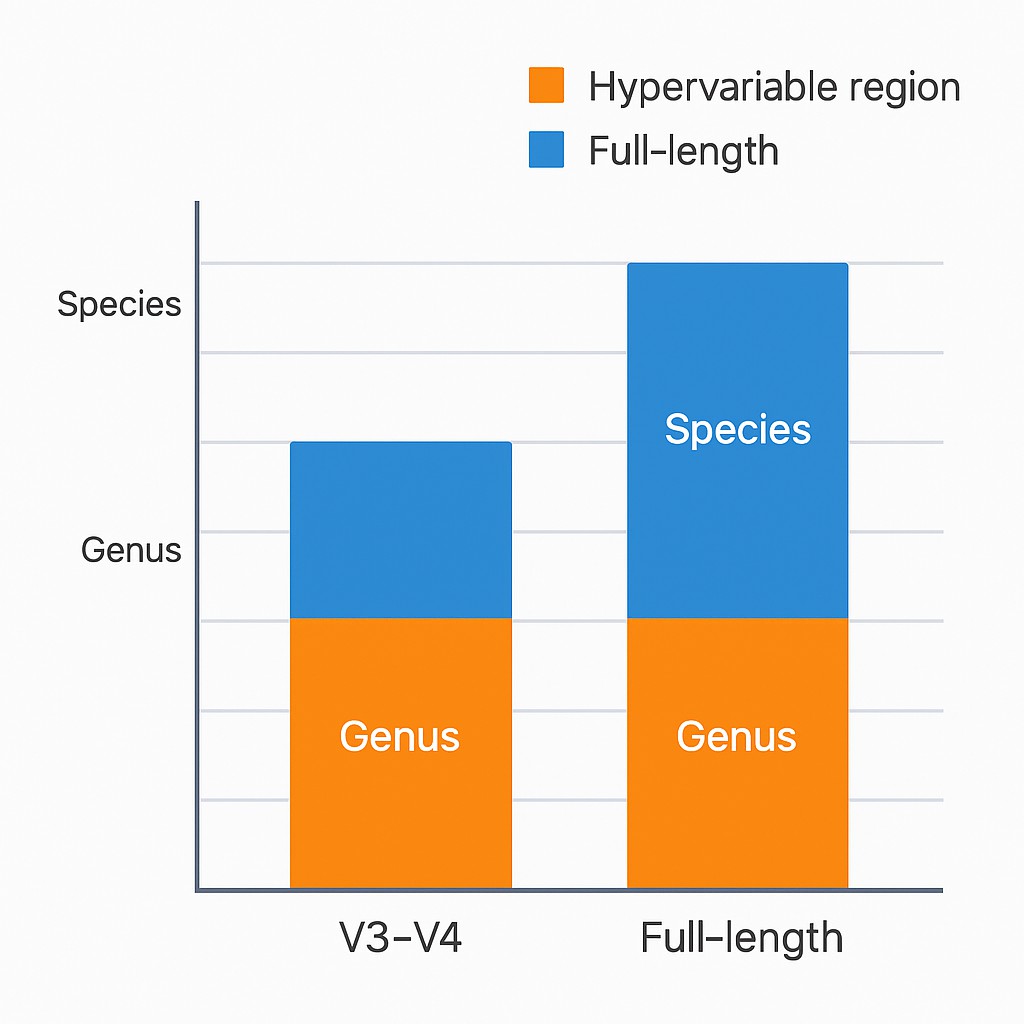
Nanopore-Amplicon-Sequenzierung verbessert die Zuordnung auf Artenebene erheblich im Vergleich zu Kurzleseverfahren.
Konsensgenauigkeit mit Fehlerunterdrückung

Fortgeschrittene Fehlerkorrektur-Workflows reduzieren falsch-positive Ergebnisse und Chimärenbildung und gewährleisten eine hochpräzise Konsensbildung.
Analyse der Gemeinschaftsvielfalt
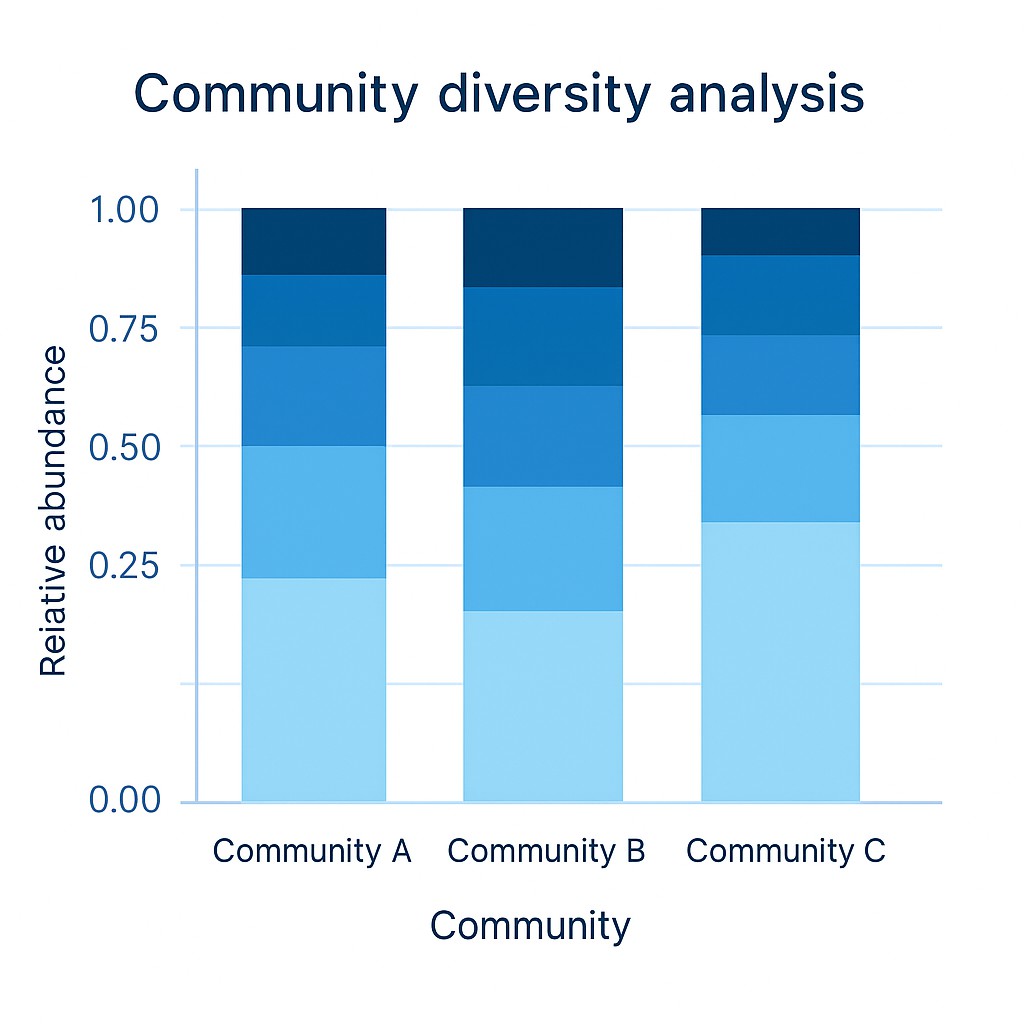
Die Beta-Diversitätsanalyse ermöglicht eine klare Trennung von mikrobiellen Gemeinschaften über die Probengruppen hinweg.
Nanopore Amplicon-Sequenzierung FAQs
Q1. Was unterscheidet die Nanopore-Amplicon-Sequenzierung von den Illumina-Short-Read-Methoden?
Nanopore liefert vollständige Lesevorgänge (z. B. vollständige 16S V1–V9 oder ITS), die eine Auflösung auf Arten- oder Stammebene ermöglichen. Illumina-Lesevorgänge sind kürzer (z. B. V3–V4) und können Arten falsch klassifizieren oder Details auf Stammebene verlieren.
Q2. Wann ist eine barcodebasierte Fehlerkorrektur (einzigartige Kennzeichnung) erforderlich?
Verwenden Sie es, wenn:
- Sie benötigen eine sehr hohe Genauigkeit (z. B. Variantenentdeckung in klonalen oder gemischten Proben),
- Zielgerichtete lange Amplicons, die anfällig für Sequenzierungs- oder PCR-Fehler sind,
- Untersuchung seltener Taxa.
Q3. Wie viele Reads pro Probe werden empfohlen?
- Für die Gemeinschaftsprofilierung: ≥ 50.000 Reads/Stichprobe, um die Vielfalt in Proben mit moderater Komplexität zu sättigen.
- Für klonale Amplicons sind weniger Reads erforderlich, da der Fokus auf der Konsensgenauigkeit und nicht auf der Vielfalt liegt.
Q4. Welche Faktoren beeinflussen die Datenqualität am stärksten?
- DNA-Eingabe: Menge, Reinheit (OD-Verhältnisse), Integrität (Fragmentgröße).
- PCR-Spezifität: saubere Einzelband-Amplifikationen.
- Probenhandhabung: Gefrier-Tau-Zyklen, Kontamination und Inhibitoren vermeiden.
Q5. Wie schnell werde ich Ergebnisse erhalten?
Die Durchlaufzeit hängt von der Projektart und der Qualität der Proben ab. Klonale Projekte sind in der Regel schneller, während Gemeinschaftsstudien aufgrund zusätzlicher Analyse-Schritte länger dauern können.
Q6. Sind die Ergebnisse für die Veröffentlichung oder die regulatorische Verwendung geeignet?
Ja, für Nur für ForschungszweckeWir liefern Veröffentlichungsberichte in hoher Qualität, Methodendetails und versionierte Taxonomiedatenbanken. Dieser Service ist jedoch nicht für Diagnosen zertifiziert.
Q7. Welche internen Referenzdatenbanken werden für die taxonomische Zuordnung verwendet?
Wir verwenden kuratierte, regelmäßig aktualisierte 16S/ITS-Volltext-Referenzdatenbanken, um eine verbesserte Zuordnung auf Artenebene und minimale Fehlklassifikationen zu gewährleisten.
Q8. Kann ich mehrere Genziele oder Loci in einer einzigen Probe sequenzieren?
Ja, vorausgesetzt, die PCR-Amplifikationen sind spezifisch (ein einzelnes, eindeutiges Produkt pro Ziel). Gemischte oder unspezifische Amplicons verringern die Genauigkeit und können separate Durchläufe oder maßgeschneiderte Arbeitsabläufe erfordern.
Nanopore-Amplikon-Sequenzierung Fallstudien
Kundenhintergrund
Forscher von TU Dortmund Universität (Deutschland) und Nationale Universität des Südens (Argentinien) untersuchte das Actinomycet Streptomyces albus CAS922. Dieser Stamm wurde aus Sonnenblumensamenhülsen isoliert und zeigte ein starkes Potenzial für Lignocellulose-Abbau und Biosynthese sekundärer MetabolitenUm seine metabolische Kapazität und industrielle Relevanz zu erkunden, benötigte das Team ein hochwertige vollständige Genomsequenz.
Herausforderung
- Aktinobakterien enthalten typischerweise große, GC-reiche Genome (>70%), was die Sequenzierung und Assemblierung erschwert.
- Kurzlesetechnologien erzeugen häufig fragmentierte Assemblierungen mit ungelösten Wiederholungen, was die nachgelagerte Analyse von biosynthetischen Genclustern (BGCs) und carbohydrate-aktiven Enzymen (CAZymes) einschränkt.
- Der Kunde benötigte einen Ansatz, der lange, zusammenhängende Reads erzeugen und eine genaue Annotation für die funktionelle Genomforschung unterstützen konnte.
Unsere Lösung
- CD Genomics führte eine Oxford Nanopore Long-Read-Sequenzierung mit dem Ligation-Kit SQK-LSK109 durch.
- Datenproduktion: Über 1,5 Gb saubere Daten aus mehr als 260.000 Reads, mit einer N50-Read-Länge von etwa 8 kb.
- Assemblierungsstrategie: De-novo-Assemblierung mit Canu, gefolgt von Politur mit Pilon zur Verbesserung der Genauigkeit.
- Annotierungswerkzeuge: NCBI Prokaryotic Genome Annotation Pipeline (PGAP) zur Genanrufung; RepeatMasker für repetitive Elemente; CAZy/dbCAN2 zur Vorhersage von carbohydrate-aktiven Enzymen; antiSMASH zur Erkennung biosynthetischer Cluster.
Ergebnisse
- Ein vollständiges lineares Chromosom von 8,06 Mb wurde assembliert, mit einer Abdeckung von etwa 191× und einem GC-Gehalt von 72,6 %.
- Es wurden 6.776 protein-codierende Gene und 80 RNAs identifiziert.
- Es wurden 232 kohlenhydrataktive Enzyme vorhergesagt, darunter drei kupferabhängige Enzyme, die an der Zellulose- und Xylanabbau beteiligt sind.
- Es wurden 27 biosynthetische Gencluster entdeckt, die Metaboliten wie Siderophore (Desferrioxamin E), Terpene (Cyslabdan, Geosmin) und Antibiotika (Xantholipin, Pseudouridimycin) kodieren.
Auswirkung
- Das hochqualitative Genom ermöglichte funktionsspezifische Einblicke auf Stamm-Ebene in den Lignocellulose-Abbau und unterstützte die Entwicklung erneuerbarer Biomasseanwendungen.
- Die Entdeckung vielfältiger biosynthetischer Cluster hat das biotechnologische Potenzial von S. albus CAS922 zur Produktion neuartiger Antibiotika und bioaktiver Verbindungen hervorgehoben.
- Das Projekt zeigte, wie der Nanopore-Sequenzierungsdienst von CD Genomics komplexe, hoch-GC-reiche Genome auflösen und umsetzbare biologische Erkenntnisse liefern kann.
Referenzen:
- Tippelt A, Nett M, Vela Gurovic MS. Vollständige Genomsequenz des lignocellulose-abbaubaren Actinomyceten Streptomyces albus CAS922. Mikrobiologische Ressourcenankündigungen. 2020 21. Mai;9(21):e00227-20. doi: 10.1128/MRA.00227-20. PMID: 32439662; PMCID: PMC7242664.
- Sonne B, Bhati KK, Song P, Edwards A, Petri L, Kruusvee V, Blaakmeer A, Dolde U, Rodrigues V, Straub D, Yang J, Jia G, Wenkel S. Die FIONA1-vermittelte Methylierung des 3'UTR von FLC beeinflusst die FLC-Transkriptspiegel und die Blüte in Arabidopsis.. PLoS Genetik2022 Sep 27;18(9):e1010386. doi: 10.1371/journal.pgen.1010386. PMID: 36166469; PMCID: PMC9543952.


