
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
5mC/5hmC-Sequenzierung
CD Genomics bietet fortschrittliche 5mC/5hmC-Sequenzierungsdienste an, um DNA-Methylierungs- und Hydroxymethylierungsmuster zu entschlüsseln. Unser hochauflösender Ansatz liefert entscheidende Einblicke in die epigenetische Regulation über Genome hinweg und unterstützt die Forschung in der Entwicklungsbiologie, den Krankheitsmechanismen und darüber hinaus. Kontaktieren Sie uns, um Ihre epigenetische Forschung noch heute voranzutreiben.
Was ist 5mC und 5hmC?
DNA-Methylierung und Hydroxymethylierung an der 5-Position von Cytosin (5mC) sind wesentliche epigenetische Veränderungen, die eine entscheidende Rolle bei der Regulierung der Genexpression und der Zell-Differenzierung spielen. DNA-Methylierung unterdrückt typischerweise die Gen-Transkription. Hydroxymethylierung hingegen aktiviert die Genexpression oder fördert die DNA-Demethylierung. 5mC-Gruppen, die durch DNA-Methyltransferasen eingeführt werden, können iterativ zu 5-Hydroxymethylcytosin (5hmC), 5-Formylcytosin (5fC) und 5-Carboxylcytosin (5caC) durch die Wirkung von Tet-Methylcytosin-Dioxygenasen oxidiert werden. 5hmC ist der am häufigsten vorkommende Bestandteil in vivo unter den drei 5mC-oxidativen Derivaten und kann in nahezu allen Säugetiergeweben und -zellen identifiziert werden. Die genomweite Kartierung von 5mC und 5hmC deckt die genomischen Bereiche dieser Veränderungen auf, was entscheidend für das Verständnis ihrer Mechanismen ist.
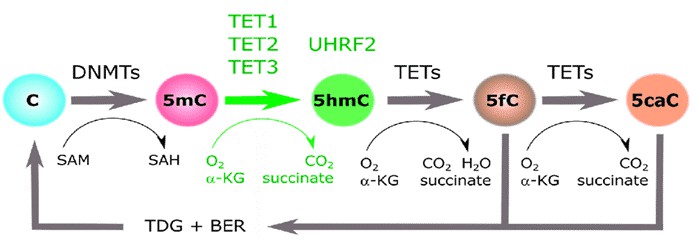 Abbildung 1. Der Cytosin-Methylierungszyklus (Ecsedi, 2018)
Abbildung 1. Der Cytosin-Methylierungszyklus (Ecsedi, 2018)
Die Einführung der 5mC/5hmC-Sequenzierung
Whole-Genome-Bisulfid-Sequenzierung (WGBS) gilt als der "Goldstandard" für Methylierungssequenzierung, der hauptsächlich die beiden Formen der Modifikation, 5mC und 5hmC, nachweist. Daraus wurden mehrere abgeleitete Methoden entwickelt, um zwischen 5mC und 5hmC zu unterscheiden. Zum Beispiel, Oxidations-Bisulfid-Sequenzierung (oxBS-seq) wird für 5mC-spezifische Sequenzierung verwendet, und die TET-unterstützte Bisulfit-Sequenzierung (TAB-seq) ist spezifisch für die Erkennung von 5hmC. In den letzten Jahren hat sich 5hmC als ein bedeutender Biomarker für das Screening von Krebs herausgestellt und zeigt einen entscheidenden klinischen Wert.
Konventionelles Bisulfite-Sequencing (BS-seq) unterscheidet nicht zwischen 5mC und 5hmC und liefert ein gemischtes Signal der beiden Modifikationen. Oxidations-Bisulfid-Sequenzierung (oxBS-seq) oxidiert 5hmC zu 5fC und wandelt es anschließend mit Bisulfitbehandlung in die Base U um, was eine präzise Erkennung von 5mC ermöglicht. Die gleichzeitige BS-Seq- und oxBS-Seq-Sequenzierung derselben Probe kann eine Erkennung von 5hmC mit Einzelbasenauflösung erreichen und bietet einen hydroxymethylierungsempfindlichen Ansatz mit Basenauflösung.
CD Genomics ist ein Biotechnologieunternehmen, das sich auf Sequenzierungstechnologie spezialisiert hat. Mit modernster Sequenzierungstechnologie und umfassender bioinformatischer Analyse unterstützen wir Kunden dabei, genomische Informationen zu erschließen. Zu unseren Fachgebieten gehören Genomik, Transkriptomik, Epigenetik, Populationsgenetik, Mikrobiologie und viele weitere Bereiche. Unser Hochdurchsatz-5mC/5hmC-Sequenzierungsdienst nutzt mehrere ausgereifte und stabile Plattformen mit hoher Effizienz, Einfachheit und Genauigkeit, um Ihre epigenetische Forschung zu unterstützen.
CD Genomics kann genomweite 5mC- und 5hmC-Detektion durch folgende Ansätze bereitstellen:
1. Antikörperbasierte Immunpräzipitation und Sequenzierung von hydroxymethylierter DNAhMeDIP-seq).
2. Oxidative Bisulfid-Sequenzierung (oxBS-seq)
3. Whole-Genome-Bisulfid-/oxidative Bisulfid-SequenzierungWGBS/oxWGBS-seq)
4. Reduzierte repräsentative Bisulfid-oxidative Bisulfid-SequenzierungRRBS/oxRRBS)
Vorteile der 5mC/5hmC Sequenzierung
- Hochauflösende Methylierungsprofilierung: Diese Methodik bietet eine detaillierte Kartierung der DNA-Methylierung (5mC) und Hydroxymethylierung (5hmC) im gesamten Genom und liefert präzise Einblicke in Methylierungsmuster, nicht nur an CpG-Stellen, sondern auch darüber hinaus.
- Quantitative Messung: Sie ermöglicht die präzise Quantifizierung von Methylierungsgraden in spezifischen genomischen Regionen wie Promotoren und CpG-Inseln und gewährt ein umfassendes Verständnis der Mechanismen, die der epigenetischen Regulation zugrunde liegen.
- Gesamtgenomabdeckung: Diese Technik umfasst das gesamte Genom und erstreckt sich auf entfernte und nicht kodierende Regionen, wodurch ein ganzheitliches Verständnis der Rolle von Methylierung bei der Genregulation und breiteren epigenetischen Phänomenen ermöglicht wird.
- Hohe Durchsatzrate und Effizienz: Gekennzeichnet durch die Fähigkeit, große Stichprobengrößen effizient zu verwalten, ist dieser Ansatz besonders gut geeignet, um komplexe Datensätze zu verarbeiten und hochdurchsatzanalysen auf eine optimierte Weise zu ermöglichen.
- Vergleichende Analyse: Durch die Unterstützung vergleichender Bewertungen von Methylierungsprofilen über mehrere Proben hinweg werden Variationen aufgedeckt, die mit unterschiedlichen physiologischen Zuständen, Krankheiten oder experimentellen Einstellungen korreliert sind, und wertvolle Einblicke in biologische Reaktionen auf epigenetischer Ebene gewonnen.
Anwendungen der 5mC/5hmC-Sequenzierung
- Krankheitsforschung und Diagnose: Identifiziert Methylierungsmarker, die mit Krankheiten assoziiert sind und potenziell als frühe diagnostische oder prognostische Biomarker bei Krebs und anderen Erkrankungen dienen können.
- Entwicklung und Differenzierung: Untersucht epigenetische Mechanismen in biologischen Entwicklungs- und Differenzierungsprozessen und erläutert die Rolle der Methylierung bei der Bestimmung des Zellschicksals und der gewebespezifischen Genexpression.
- Umweltreaktion und genetische Variation: Analysiert, wie Umweltfaktoren die Methylierungsmuster beeinflussen und untersucht die Beziehung zwischen genetischen Variationen und epigenetischen Veränderungen, die Einblicke in die Umweltanpassung und phänotypische Plastizität bieten.
- Arzneimittelentwicklung und therapeutische Strategien: Bewertet die Auswirkungen von Kandidatenmedikamenten auf Methylierungsmuster und trägt zur Entdeckung neuer therapeutischer Ziele und Strategien bei.
- Agrarwissenschaften und Pflanzenbiologie: Untersucht Methylierungsmuster in Pflanzengenomen, um deren Rolle in der Pflanzenzüchtung, der Verbesserung der Stressresistenz und der landwirtschaftlichen Produktivität zu verstehen.
5mC/5hmC Sequenzierungs-Workflow
Unsere langjährige Erfahrung und fortschrittlichen Plattformen ermöglichen es uns, ein umfassendes Servicepaket von der Projektberatung über das Experimentdesign, die Sequenzierung bis hin zur bioinformatischen Analyse anzubieten. Wir bemühen uns, das geeignetste Konzept für Ihr Projekt bereitzustellen.
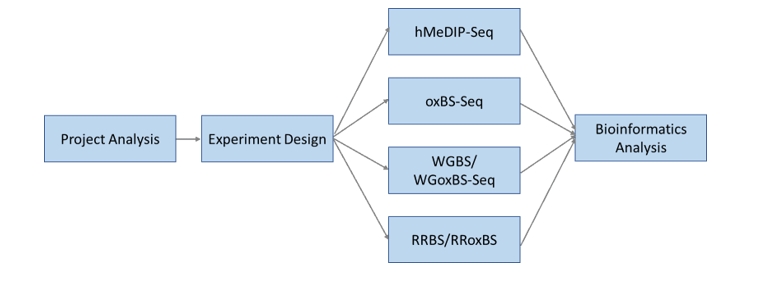
Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur 5mC/5hmC-Sequenzierung für Ihr Schreiben (Anpassung)
Erforschen Sie DNA-Methylierung und Hydroxymethylierung mit dem 5mC/5hmC-Sequenzierungsdienst von CD Genomics. Wir bieten hochauflösende Sequenzierung und umfassende bioinformatische Analysen, um epigenetische Muster im gesamten Genom aufzudecken. Kontaktieren Sie uns, um Erkenntnisse für Ihre Forschung zu gewinnen.
Referenzen
- Ecsedi S, Rodríguez-Aguilera JR, Hernandez-Vargas H. 5-Hydroxymethylcytosin (5hmC), oder wie man seine Lieblingszelle identifiziert. Epigenome2018, 2(1):3.
- Kirschner K, Krueger F, Green AR, Chandra T. Multiplexing für oxidative Bisulfite-Sequenzierung (oxBS-seq). In DNA-Methylierungsprotokolle 2018. Humana Press.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

5mC/5hmC-Sequenz FAQs
1. Wie beeinflusst 5hmC die Gentranskription?
Diese epigenetische Modifikation, 5-Hydroxymethylcytosin (5hmC), spielt eine entscheidende Rolle bei der Gentranskription, indem sie die Chromatinstruktur moduliert und die Rekrutierung spezifischer regulatorischer Proteine orchestriert. Ihre Assoziation mit aktiven transkriptionalen Zuständen deutet auf ihre Fähigkeit hin, die Prozesse der Genexpression zu erleichtern. Bemerkenswerterweise stört 5hmC die Dynamik der DNA-Methylierung, indem es als Vermittler in Demethylierungsprozessen fungiert und somit die genetischen Regulationsnetzwerke formt.
2. Warum ist 5hmC bedeutend?
Seine Rolle als dynamischer Marker für Veränderungen der DNA-Methylierung unterstreicht seinen wesentlichen Beitrag zur Regulierung kritischer biologischer Prozesse wie Entwicklung, Differenzierung und Krankheitsprogression. Im Gegensatz zu den repressiven Effekten von 5-Methylcytosin (5mC) wird 5hmC aktivierenden transkriptionalen Aktivitäten und regulatorischen Funktionen im Genom zugeschrieben. Das Entschlüsseln der komplexen Muster von 5hmC bietet entscheidende Einblicke in die epigenetischen Mechanismen, die normale physiologische Funktionen und pathologische Zustände steuern, und birgt das Potenzial für diagnostische und therapeutische Fortschritte bei Erkrankungen wie Krebs und neurologischen Störungen.
3. Wie unterscheidet sich 5hmC von 5mC in Bezug auf die biologische Funktion?
Im Bereich der Derivate der DNA-Methylierung ergibt sich ein bemerkenswerter Unterschied zwischen 5hmC und 5mC. Während beide Moleküle an dieser epigenetischen Modifikation beteiligt sind, hebt sich 5hmC durch seine Verbindung zur aktiven Transkription und seine Rolle in den komplexen Prozessen der DNA-Demethylierung hervor. Bemerkenswert ist, dass sein Einfluss auf die Dynamik der Genexpression und die Entwicklungswege im krassen Gegensatz zur traditionellen Rolle von 5mC steht, das für die Genstilllegung verantwortlich ist.
4. Wie kann die Sequenzierung von 5mC/5hmC zur personalisierten Medizin beitragen?
Die Nutzung der 5mC/5hmC-Sequenzierung eröffnet Möglichkeiten zur Entdeckung epigenetischer Marker, die mit der Anfälligkeit für Krankheiten, der Prognose und der Reaktionsfähigkeit auf Behandlungen verbunden sind. Dieser innovative Ansatz verspricht, personalisierte diagnostische und therapeutische Interventionen zu gestalten und damit die Landschaft der präzisen Medizin zu revolutionieren.
5mC/5hmC-Sequenz-Fallstudien
Integrierte Analysen von Multi-Omics zeigen globale Muster der Methylierung und Hydroxymethylierung und untersuchen die tumorunterdrückenden Rollen von HADHB beim kolorektalen Krebs.
Journal: Klinische Epigenetik
Impact Faktor: 7,259
Veröffentlicht: 02. März 2018
Hintergrund
DNA-Methylierung ist eine entscheidende epigenetische Modifikation, die mit der Genexpression in Verbindung steht. 5mC und 5-hmC dienen als zwei epigenetische Marker, die an der Aufrechterhaltung der epigenetischen Reprogrammierung beteiligt sind, wobei die Genom-Methylierung potenziell zu Krebs führen kann. Ziel dieser Studie ist es, die epigenetischen Mechanismen der DNA-Methylierung und Hydroxymethylierung bei der Entwicklung von kolorektalem Krebs zu erläutern. Die Autoren verwendeten MeDIP-seq und hMeDIP-seq um die DNA-Methylierungs- und Hydroxymethylierungsmuster des gesamten Genoms von kolorektalen Krebsgeschwülsten und den entsprechenden normalen Geweben zu kartieren, zusammen mit der Analyse der Transkriptom-GenexpressionRNA-SeqDifferenzielle Methylierungsregionen (DMRs), differenzielle Hydroxymethylierungsregionen (DhMRs) und differentielle exprimierte Genregionen (DEGs) wurden identifiziert. Epigenetische Biomarker wurden durch DMR-, DhMR- und DEG-Analysen ausgewählt, gefolgt von einer funktionalen Analyse zur Validierung.
Materialien & Methoden
Probenvorbereitung
- Kolorektale Tumorproben
- Benachbarte normale Gewebe
- DNA-Extraktion
- RNA-Extraktion
Sequenzierung
- RNA-Seq
- MeDIP-seq
- hMeDIP-seq
- Quantitative PCR
- Analyse des Genexpressionsniveaus
- Differenzielle Genexpressionsanalyse
- Identifizierung von DMR und DhMR
- Funktionelle Anreicherungsanalyse von DMR und DhMR
- RNA-Interferenz-Analyse
- Western-Blot-Analyse
Ergebnisse
Die Autoren untersuchten die unterschiedlichen ganzgenomischen Hydroxymethylierungsmuster, die als epigenetische Biomarker im normalen kolorektalen Gewebe dienen können. Durch den Vergleich von kolorektalen Tumor- und Normalgeweben wurden insgesamt 59.249 DMRs, 187.172 DhMRs und 948 DEGs identifiziert. Bei der Kreuzreferenzierung der Gene aus DMRs oder DhMRs mit DEGs wurden sieben Gene ausgewählt, die durch DNA-Methylierung in Tumoren aberrant reguliert waren. Darüber hinaus wurde eine hohe Methylierung des HADHB-Gens festgestellt, die mit seiner Herunterregulierung in der Transkription von kolorektalem Krebs (CRC) korreliert war. Die funktionelle Analyse des Tumors zeigte, dass das HADHB-Gen die Migration und Invasivität von Krebszellen reduzierte. Die Ergebnisse deuten darauf hin, dass HADHB als Tumorsuppressor-Gen (TSG) wirken könnte.
 Abb. 1 Globale Muster des Methyloms und Hydroxymethyloms.
Abb. 1 Globale Muster des Methyloms und Hydroxymethyloms.
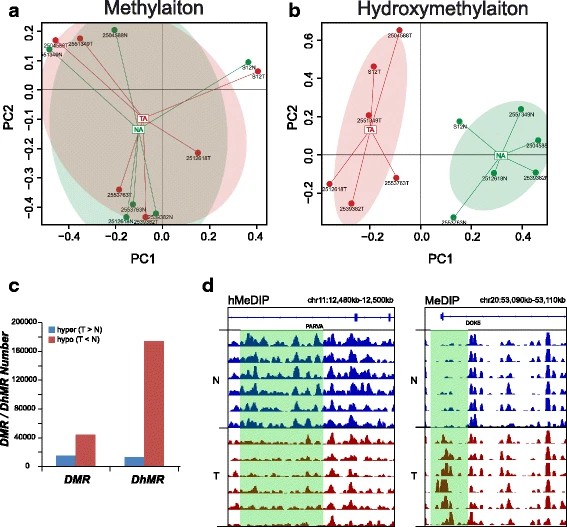 Abb. 2 Verschiedene Verteilungen von Methylierung und Hydroxymethylierung.
Abb. 2 Verschiedene Verteilungen von Methylierung und Hydroxymethylierung.
 Abb. 3 Zusammenhang zwischen epigenomischer Modifikation und Genexpression.
Abb. 3 Zusammenhang zwischen epigenomischer Modifikation und Genexpression.
Fazit
Die Studie erläutert die genomweiten Methylierungs- und Hydroxymethylierungsmuster bei CRC. Durch den Einsatz von Multi-Omics-Analysen wurden sieben mit CRC verbundene Gene identifiziert. Funktionale Experimente unterstützten die Annahme, dass HADHB als potenzielles Tumorsuppressorgen wirkt, indem es die Invasivität und Migration von Tumorzellen reduziert. Die Stummschaltung des HADHB-Gens könnte den Beginn und die Progression von kolorektalem Krebs fördern.
Referenz
- Zhu Y, Lu H, Zhang D, et al. Integrierte Analysen von Multi-Omics zeigen globale Muster der Methylierung und Hydroxymethylierung und untersuchen die tumorunterdrückenden Rollen von HADHB bei kolorektalem Krebs. Klinische Epigenetik. 2018, 10:1-3.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Journal: Naturkommunikationen
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nucleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


