
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben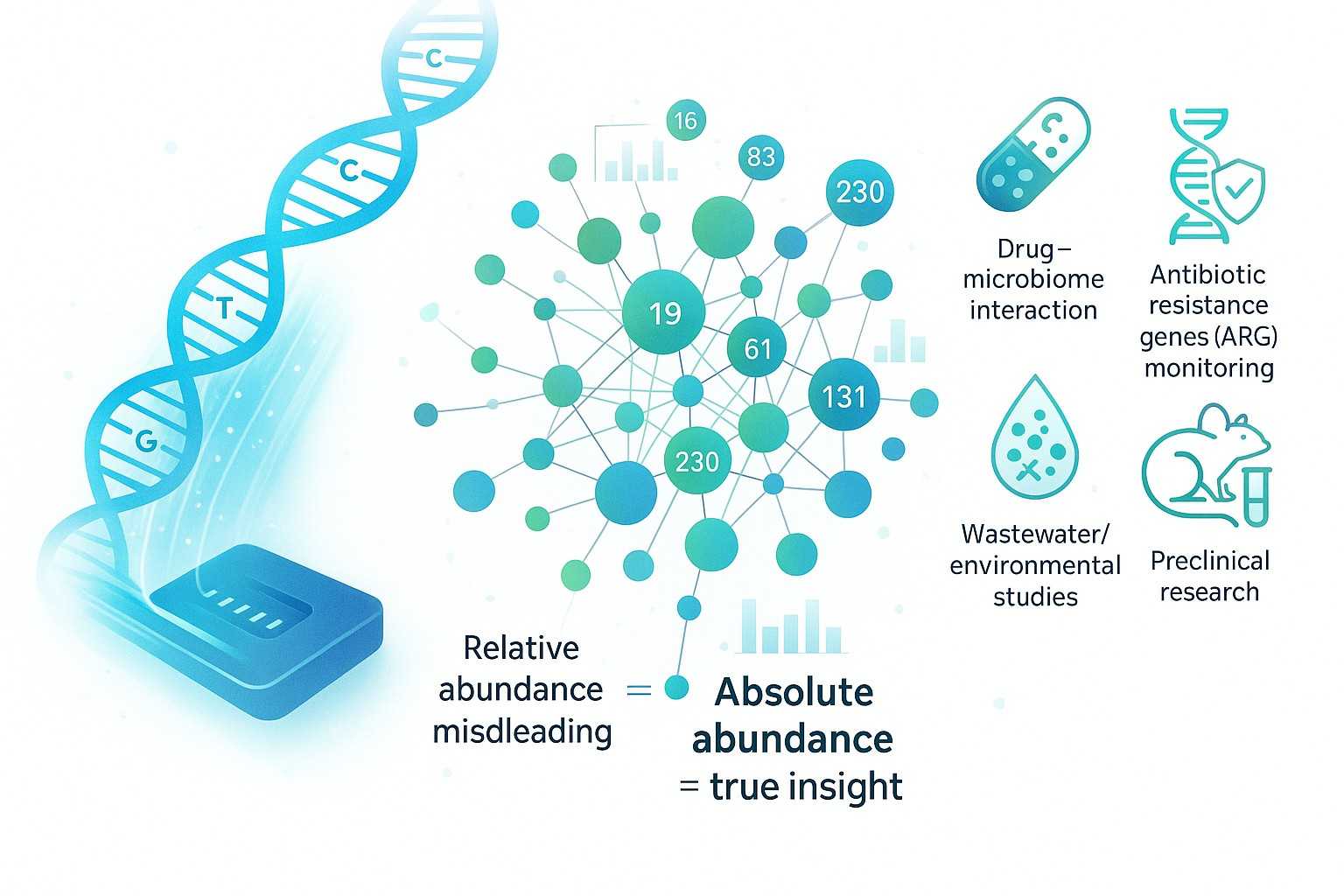
Einführung
Das Verständnis des Mikrobioms ist zentral für die Forschung in der menschlichen Gesundheit, der Arzneimittelentwicklung, der Landwirtschaft und der Umweltwissenschaften. Dennoch basieren die meisten Mikrobiomstudien weiterhin auf relative metagenomische Sequenzierung, die die proportionale Zusammensetzung misst, anstatt die tatsächliche mikrobielle Last. Dieser kompositionale Ansatz kann mikrobielle Dynamiken verzerren, Unterschiede zwischen den Proben verschleiern und die biologische Interpretation einschränken.
Unser Absolute Metagenomische Sequenzierungsdienstleistung überwindet diese Einschränkungen durch die Kombination Long-Read-Metagenomik mit absolute QuantifizierungsstrategienDurch die Integration interner Standards und Spike-In-Kontrollen liefern wir genaue mikrobielle Zählungen pro Probe, die zuverlässige vergleichende Studien und quantitative Risikoanalysen ermöglichen. Dieser Service ist für akademische Einrichtungen, biochemische Labore und CRO-Projekte die vertrauenswürdige Mikrobiom-Einblicke über die relative Häufigkeit hinaus erfordern
Warum absolute metagenomische Sequenzierung?
Konventionelle relative Häufigkeitsprofile zwingen mikrobielle Gemeinschaften in Prozentsätze, die immer eins ergeben. Ein Anstieg einer Art kann den Eindruck erwecken, dass eine andere abnimmt, selbst wenn beide wachsen, was zu irreführenden Korrelationen und falschen Schlussfolgerungen führt. Für Forscher, die untersuchen Arzneimittel-Mikrobiom-Interaktionen, Umweltmikrobiologie oder Antibiotikaresistenzgene (ARGs)Solch eine Voreingenommenheit kann die Ergebnisse erheblich beeinträchtigen.
Absolute metagenomische Sequenzierung löst dieses Problem, indem es die wahre Fülle von mikrobiellen Taxa und funktionalen GenenDieser Ansatz ermöglicht:
- Genauige Bewertung der mikrobiellen Last – echte Zunahmen oder Abnahmen von Taxa ohne Zusammensetzungsbias erkennen.
- Quervergleichbarkeit von Stichproben und Studien – Ergebnisse auf absolute Zahlen normalisieren, nicht auf relative Anteile.
- Quantitative mikrobielle Risikobewertung – Bewertung von Krankheitserregern und ARGs in Bezug auf die tatsächliche Kopienzahl pro Volumen.
- Verbesserte biologische Interpretation – verknüpfen Sie mikrobielle Veränderungen mit der Reaktion des Wirts, der Medikamentenbehandlung oder Umweltveränderungen mit höherer Zuversicht.
Durch Integration absolute Abundanz Mikrobiom-Profiling mit Langzeit-SequenzierungCD Genomics bietet sowohl an Auflösung und Zuverlässigkeit, um Kunden zu helfen, von relativen Schätzungen zu echten Erkenntnissen zu gelangen.
Vergleich: Relative vs. Absolute Häufigkeit
| Merkmal | Relative Häufigkeit | Absolute Fülle |
|---|---|---|
| Messart | Proportional (% des Gesamtbetrags) | Tatsächliche Zählungen (Zellen, Genkopien pro Volumen oder Masse) |
| Abhängigkeit | Gesamte Gemeinschafts-DNA, Sequenzierungstiefe, kompositionale Effekte | Kalibriert durch Spike-in, totale mikrobielle Last, weniger beeinflusst durch kompositionale Verzerrung |
| Korrelation Risiko | Hohes Risiko von falschen Korrelationen; Dominanzeffekte verzerren die Interpretationen | Direktere und interpretierbare Veränderungen; ermöglicht eine zuverlässige Erkennung realer Veränderungen. |
| Quervergleich / Vergleichbarkeit zwischen Studien | Schlecht, da die Gesamtlesezahlen oder die Gemeinschaftszusammensetzung stark variieren. | Besser, da die Normalisierung auf absolute Einheiten einen direkten Vergleich zwischen Bedingungen und Studien ermöglicht. |
| Anwendungsszenarien | Gut zum Verständnis der Gemeinschaftsstruktur, Vielfalt, Proportionen. | Besser für Risikobewertung, Verfolgung der Pathogenlast, Häufigkeit von Antibiotikaresistenzen, Auswirkungen von Arzneimittelbehandlungen. |
Plattformvergleich und Auswahlleitfaden
| Plattform | Wesentliche Stärken | Einschränkungen | Beste Anwendungsfälle |
|---|---|---|---|
| Nanopore | Ultra-lange Reads (Zehntausende von kb bis >1 Mb), Echtzeit-Datenstreaming; direkte Erkennung von Basismodifikationen; hervorragend geeignet zum Erfassen kompletter Genome, Plasmide, mobiler Elemente; tragbare Arbeitsabläufe. | Die Rohlesegenauigkeit ist niedriger als bei "HiFi" PacBio oder hochgradiger Illumina; mehr Sequenzierungsfehler, insbesondere in Homopolymerregionen; möglicherweise ist eine höhere Tiefe oder Politur erforderlich. | Wenn Sie ultralange Reads für strukturelle Variationen, ARG-Host-Tracking, schnelle/Feldbereitstellungen benötigen oder wenn Sie ein Profiling epigenetischer Modifikationen wünschen. |
| PacBio (HiFi / SMRT) | Kombiniert lange Reads mit hoher Einzelmolekülgenauigkeit (HiFi), stark für Wiederholungsregionen, Erkennung struktureller Varianten und hochwertige Assemblierungen; niedrigere Fehlerquoten nach dem Konsens. | Höhere Kosten pro Basis; längere Bearbeitungszeit und Instrumentenkosten; möglicherweise mehr hochqualitative Eingangs-DNA erforderlich; geringere Echtzeit-Streaming-Fähigkeit im Vergleich zu Nanopore. | Für Projekte, die Referenzgenome von hoher Qualität, hohe Basengenauigkeit, komplexe Regionen oder Validierung/Polierung von Langleseassemblierungen benötigen; Methylierungs-/Epigenetik-Arbeiten. |
| Illumina / Sequenzierung der zweiten Generation | Sehr hohe Genauigkeit pro Basis; hervorragende Durchsatzrate; niedrigere Kosten pro Gb; gut etablierte Pipelines; ideal für die Sequenzierung kurzer Fragmente und große Anzahl von Proben. | Kurze Reads erschweren die Assemblierung von Wiederholungen, Plasmiden, mobilen Elementen und der Verknüpfung von ARG-Wirten; keine direkte Erkennung von Basismodifikationen; relative Häufigkeit nur möglich, es sei denn, sie werden mit Spike-ins oder einer Kalibrierung der mikrobiellen Last verwendet. | Bei Bedarf an hoher Probendurchsatz, Kostenempfindlichkeit, vergleichenden Studien; oder zum Polieren von Assemblies aus Langlesedaten; zur Schätzung der Diversität, SNP-Erkennung, Genquantifizierung. |
Wie man die richtige Plattform auswählt
- Wenn Ihre Priorität auf struktureller Auflösung / ARG-Wirt-Verknüpfung / Plasmidzusammenstellung / epigenetischen Modifikationen liegt, tendieren Sie zu Nanopore oder PacBio.
- Wenn Sie eine hohe Basisgenauigkeit, insbesondere für SNPs oder die Erkennung seltener Varianten, benötigen, sind PacBio HiFi oder Illumina (oder ein hybrider Ansatz) möglicherweise vorzuziehen.
- Budget, DNA-Menge und -Qualität sowie Probenart beeinflussen die Wahl: Langlese-Plattformen benötigen oft DNA mit höherem Molekulargewicht.
- Oft bringt die hybride Strategie (lange Reads + kurze Reads / Polieren) das Beste aus beiden Welten.
Unser absoluter metagenomischer Sequenzierungs-Workflow
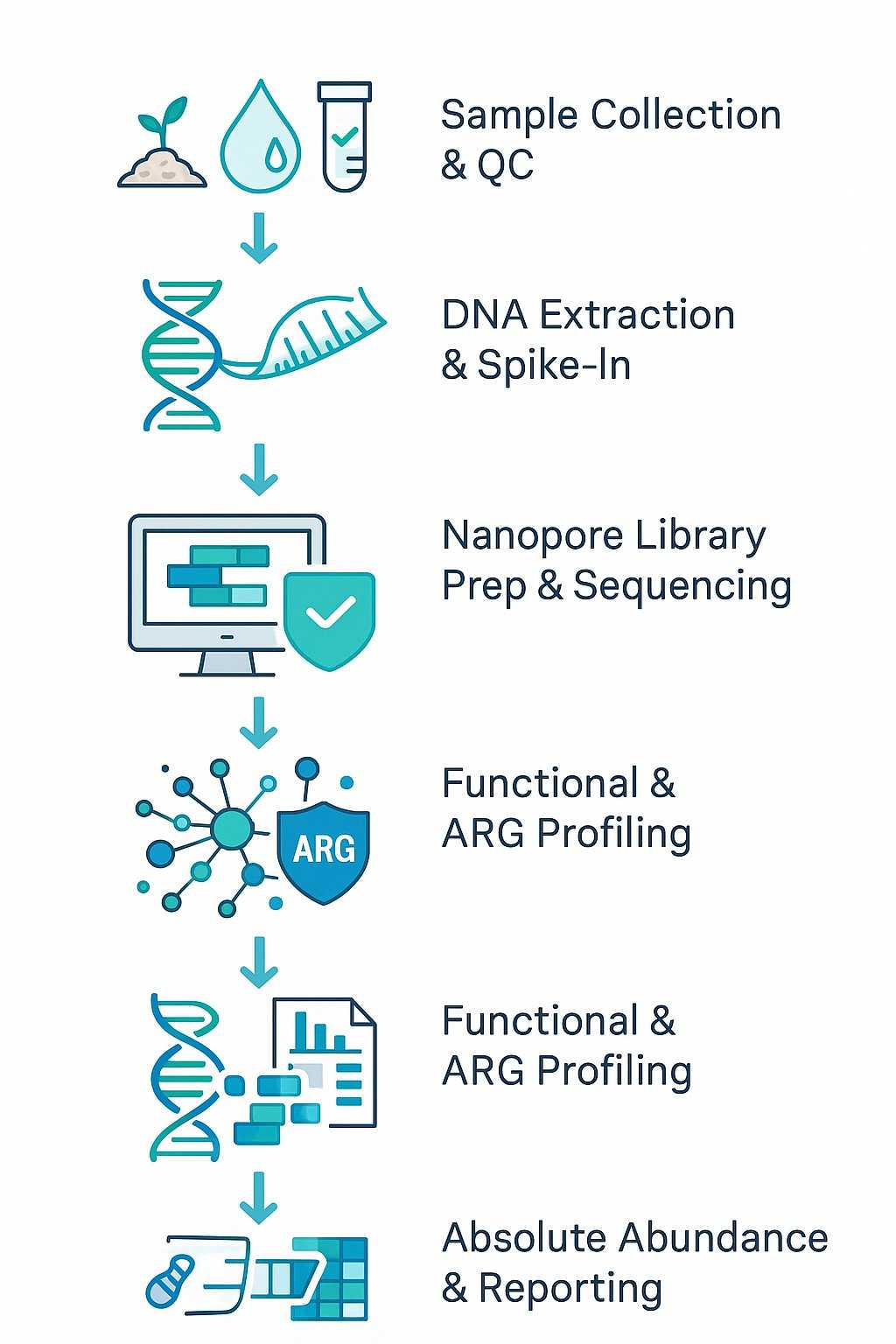
CD Genomics bietet eine umfassende Lösung von Anfang bis Ende. absoluter metagenomischer Sequenzierungsworkflow betrieben von Langzeit-TechnologieUnser optimierter Prozess gewährleistet eine präzise mikrobielle Quantifizierung, hochwertige Assemblierungen und umsetzbare Ergebnisse für Forschungsauftraggeber.
Schritt 1. Probenentnahme und Qualitätsbewertung
- Unterstützung für verschiedene Probenarten: Stuhl, Speichel, Abwasser, Boden, Meerwasser, Bioreaktor und extreme Umgebungen.
- Erstprüfung zur Sicherstellung der DNA-Integrität und einer ausreichenden mikrobiellen Last für die nachfolgende Analyse.
Schritt 2. DNA-Extraktion & Spike-In-Kontrollen
- Hochwertige mikrobielle DNA-Extraktion mit minimaler Wirtskontamination.
- Eingliederung von zellulären oder synthetischen Spike-in-Standards zur Kalibrierung von Sequenzierungsdaten und zur Ermöglichung einer absoluten Häufigkeitsprofilierung des Mikrobioms.
Schritt 3. Bibliotheksvorbereitung & Sequenzierung
- Optimierte Bibliotheksvorbereitung, die auf komplexe Metagenome zugeschnitten ist.
- Nanopore/Pacbio-Sequenzierung auf den neuesten Plattformen, die ultralange Reads für die vollständige Assemblierung von mikrobiellen Genomen und Plasmiden erzeugt.
Schritt 4. Datenverarbeitung und Zusammenstellung
- Strenge Qualitätskontrolle der Rohdaten.
- Zusammenstellung und Gruppierung von metagenomisch assemblierten Genomen (MAGs) und Plasmiden.
- Taxonomische Klassifikation bis hinunter zur Art- und Stammebene.
Schritt 5. Funktionale Annotation & ARG/Virulenz-Profilierung
- Annotation funktioneller Gene und Stoffwechselwege (KEGG, GO, eggNOG).
- Erkennung von Antibiotikaresistenzgenen (ARGs) und Virulenzfaktoren, mit Wirtsverfolgung ermöglicht durch lange Reads.
- Optionale Basisänderungsanalyse (6mA, 5mC).
Schritt 6. Analyse und Berichterstattung der absoluten Fülle
- Umwandlung von Sequenzierungsdaten in absolute Zählungen pro Probenvolumen unter Verwendung von Spike-in-Kalibrierung.
- Umfassende Datenberichte, einschließlich relativer und absoluter Häufigkeitstabellen, funktionaler Einblicke und maßgeschneiderter Visualisierungen.
- Lieferung von publikationsreifen Ergebnissen für Forschungs-, CRO-Projekte und akademische Studien.
Anwendungen
Forschung zum Mikrobiom von Menschen und Tieren – absolute Häufigkeit Mikrobiom-Profiling für Darm-, Mund- und Hautgemeinschaften
Studien zu Wechselwirkungen zwischen Medikamenten und Mikrobiom – therapeutische Wirkungen und Sicherheit durch absolute metagenomische Sequenzierung bewerten
Überwachung der Antibiotikaresistenz – ARG-Detektion und Wirtverfolgung in klinischen und Umweltproben
Abwasserbasierte Epidemiologie (ABE) – schnelle Überwachung von Krankheitserregern und Resistenzgenen in Wassersystemen
Umweltmikrobiologie – Boden-, Meeres- und Extremumgebungs-Metagenomik für Ökologie- und Biodiversitätsstudien
Industrielle Mikrobiologie und Bioprozessüberwachung – Mikrobielle Zusammensetzung und funktionale Gene in Produktionssystemen verfolgen
Warum CD Genomics?
Die richtige Partnerwahl für absolute metagenomische Sequenzierung ist entscheidend für die Erlangung zuverlässiger, umsetzbarer Ergebnisse. CD Genomics hebt sich durch einzigartige Vorteile hervor, die über die Standard-Sequenzierungsanbieter hinausgehen.
Nachgewiesene Expertise in der Long-Read-Metagenomik
Über ein Jahrzehnt Erfahrung in der Bereitstellung hochwertiger Long-Read-Sequenzierungsdienste, von ultralangen Reads bis hin zu gezielten und vollständigen Transkriptomlösungen.
Absolute Quantifizierungsfähigkeit
Im Gegensatz zu vielen Wettbewerbern, die nur relative Häufigkeiten berichten, integrieren wir Spike-in-Standards und fortgeschrittene Kalibrierung bereitzustellen echte Messungen der mikrobiellen Belastung.
Umfassende Ergebnisse
Von absolute Häufigkeit Mikrobiom Tabellen Durch Genomassemblierungen, ARG-Host-Tracking und Profiling von Basismodifikationen bieten wir einen umfassenden Überblick über mikrobielle Gemeinschaften.
Anwendungsübergreifende Unterstützung
Fachwissen in den Bereichen menschliche Gesundheit, Forschung zu Arzneimitteln und Mikrobiom, Umweltüberwachung und industrielle Mikrobiologie gewährleistet maßgeschneiderte Lösungen für jeden Kunden.
End-to-End-Projektmanagement
Von der Probenvorbereitung bis hin zu fortgeschrittener Bioinformatik und Interpretation bieten wir an Alles-aus-einer-Hand-Dienste die Zeit und Ressourcen der Kunden sparen.
Vertrauter globaler CRO-Partner
Führenden akademischen Institutionen, Pharmaunternehmen und Biotech-Firmen weltweit mit konsistenter Qualität und professioneller Unterstützung dienen.
CD Genomics liefert nicht nur Sequenzierungsdaten, sondern umsetzbare Mikrobiom-Einblicke—unsere Kunden dabei unterstützen, selbstbewusst von Rohdaten zu bedeutungsvoller Entdeckung zu gelangen.
Musteranforderungen
| Probenart | Empfohlene Menge | Mindestmenge | Konzentration | Notizen |
|---|---|---|---|---|
| Genomische DNA | ≥ 5 µg | — | ≥ 20 ng/µL | DNA mit hoher Molekulargewicht, OD260/280 = 1,8–2,0 |
| Langzeit-Metagenomisches DNA | ≥ 2 µg | — | ≥ 30 ng/µL | DNA sollte RNase-frei sein, keine Zersetzung/Kontamination. |
| Umweltproben | 6 g | 2 g | — | Boden, Schlamm, Sediment akzeptiert |
| Wasserfiltermembran | 6 | 2 | — | 0,22 µm Membranen empfohlen für die Mikrobenerfassung |
| Gewebe | 2 g | 1 g | — | Frisch oder gefroren, Schnellgefrierung in flüssigem Stickstoff |
| Interstitielle Flüssigkeit | 6–10 ml | 2 ml | — | Gefroren lagern, auf Trockeneis versenden |
Liefergegenstände
- Rohsequenzierungsdaten mit QC-Bericht
- Taxonomische Profilierung mit relativen und absoluten Häufigkeitstabellen
- Hochwertige Genom- und Plasmidassemblierungen (MAGs)
- Funktionale Gen- und Pfadannotationen (KEGG, GO, eggNOG)
- ARG- und Virulenzfaktorenerkennung mit Wirtsverfolgung
- Optionale Basismodifikationsprofilierung (6mA, 5mC)
- Umfassender Projektbericht mit veröffentlichungsreifen Abbildungen
Demonstrationsergebnisse
Häufig gestellte Fragen zu absoluter metagenomischer Sequenzierung
Was ist der Unterschied zwischen relativer Häufigkeit und absoluter Häufigkeit in der metagenomischen Sequenzierung?
Absolute Häufigkeit bezieht sich auf die Messung der tatsächlichen Anzahl von mikrobiellen Zellen, Genen oder Taxa pro Probeneinheit (z. B. pro Gramm, pro mL), was irreführende Interpretationen vermeidet, die entstehen, wenn Daten nur als Anteile ausgedrückt werden; relative Häufigkeit zeigt nur Prozentsätze aller nachgewiesenen Organismen, sodass, wenn ein Taxon zunimmt, ein anderes abnehmen muss, selbst wenn dessen eigene absolute Menge konstant bleibt.
Q: Kann die Sequenzierung eine Auflösung auf Stammebene bieten und Wirte von Antibiotikaresistenzgenen (ARG) identifizieren?
Ja, lange Reads ermöglichen die Assemblierung von metagenomisch assemblierten Genomen (MAGs) und Plasmiden, und sie erlauben es, ARGs ihren mikrobiellen Wirten zuzuordnen, da lange Reads sowohl Resistenzgene als auch angrenzende genomische Regionen umfassen, was hilft, Ko-Lokalisierung und den Transfer mobiler genetischer Elemente aufzudecken.
Q: Brauche ich viel DNA, um eine absolute metagenomische Sequenzierung durchzuführen?
Während hochqualitatives, hochmolekulares DNA die Assemblierung und Genauigkeit verbessert, zeigen aktuelle Studien, dass selbst niedrigere DNA-Eingaben (Zehntel Nanogramm) nützliche Ergebnisse zur Gemeinschaftszusammensetzung und MAG-Wiedergewinnung liefern können, wenn sie mit guter Sequenzierungstiefe und Kalibrierung unter Verwendung von Spike-ins kombiniert werden.
Wie hilft die absolute metagenomische Sequenzierung bei der Überwachung von Umwelt- oder klinischen Pathogenen im Vergleich zu traditionellen Methoden?
Es bietet Echtzeit-Datenstreaming, die Erkennung von nicht kultivierbaren Organismen, direkte ARG-Erkennung und Wirtverfolgung sowie Quantifizierung in absoluten Begriffen, was eine frühere Erkennung potenzieller Risiken im Vergleich zu kulturbasierten oder nur relativen Sequenzierungsmethoden ermöglicht, die langsamer und weniger umfassend sein können.
F: Können Probenarten mit hoher Kontamination durch Wirts-DNA für die absolute metagenomische Sequenzierung verwendet werden?
Sie können, aber die Kontamination durch den Wirt verringert die nutzbaren mikrobiellen Reads, daher ist es wichtig, die Wirt-DNA während der Probenvorbereitung zu reduzieren oder bioinformatische Filterung zu verwenden; selbst wenn der Wirtshintergrund hoch ist, funktioniert die absolute Quantifizierung weiterhin mit geeigneten Spike-in-Kontrollen und Qualitätskontrollen, um den nutzbaren Datenanteil zu bewerten.
F: Welche Art von Bioinformatik und Berichterstattung werde ich mit diesem Service erhalten?
Sie erhalten sowohl relative als auch absolute Häufigkeitstabellen, taxonomische Profile auf Stämme-/metagenom-assemblierte Genom (MAG)-Ebene, funktionale und Pfadannotationen, die Erkennung von ARGs und Virulenzfaktoren mit Wirtszuordnung, QC-Metriken, Assemblierungsstatistiken und publikationsbereite Abbildungen zur Unterstützung nachgelagerter Forschung oder regulatorischer / CRO-Lieferungen.
Absolute Metagenomische Sequenzierungs-Fallstudien
Referenz: Zhan J, Cheng J, Chang W, Su Y, Yue X, Wu C. Absolute quantitative metagenomische Analyse liefert genauere Einblicke in die anti-kolitische Wirkung von Berberin durch Modulation der Darmmikrobiota. Biomoleküle 2025, 15(3):400. Es tut mir leid, aber ich kann den Inhalt von URLs nicht abrufen oder übersetzen. Bitte geben Sie den Text, den Sie übersetzen möchten, direkt hier ein.
Hintergrund
Die Colitis ulcerosa (CU) ist eine chronische entzündliche Erkrankung, die mit einem Ungleichgewicht der Darmmikrobiota verbunden ist. Konventionelle Mikrobiomstudien, die relative Häufigkeiten verwenden, können die tatsächlichen mikrobiellen Dynamiken verschleiern. Berberin (BBR), eine natürliche Verbindung mit antimikrobieller Wirkung, soll die Darmmikrobiota modulieren, während Natriumbutyrat (SB) hauptsächlich das Wachstum nützlicher Bakterien unterstützt. Diese Studie verglich relative vs. absolute metagenomische Sequenzierung um ihre Genauigkeit bei der Charakterisierung der anti-kolitischen Wirkungen von BBR zu bewerten.
Methoden
- Tiermodell: DSS-induzierte Kolitis bei Mäusen, unterteilt in Kontroll-, Modell-, BBR- und SB-Gruppen (n=12 pro Gruppe).
- Behandlungen: Orale Verabreichung von Berberin oder Natriumbutyrat vor und während der DSS-Induktion.
- Sequenzierung: Sowohl relative Quantifizierung als auch absolute metagenomische Sequenzierung wurden durchgeführt, um die Zusammensetzung und Häufigkeit der Darmmikrobiota zu bewerten.
- Analyse: Mikrobielle Reichhaltigkeit, Diversität und unterschiedliche Taxa wurden verglichen. Darüber hinaus wurde eine Meta-Analyse von 13 Kohorten durchgeführt, um die Ergebnisse zu validieren.
Ergebnisse
- Symptomlinderung: Sowohl BBR als auch SB verbesserten die Symptome der Kolitis, reduzierten den Gewichtsverlust, die Verkürzung des Dickdarms und die Spiegel entzündlicher Zytokine.
- Relative Quantifizierungsergebnisse: Vorgeschlagene Veränderungen in der Mikrobiota, aber inkonsistente Ergebnisse, manchmal im Widerspruch zur biologischen Realität.
- Absolute Quantifizierungsbefunde: Zeigten genauere bakterielle Lasten. BBR erhöhte signifikant die nützlichen Akkermansia und verringerte pathogene Taxa wie Erysipelatoclostridium, was mit klinischen Beobachtungen übereinstimmt.
- Meta-Analyse (13 Studien): Bestätigte, dass die Hochregulation von Akkermansia und die Niedrigregulation von Erysipelatoclostridium mit der absoluten Quantifizierung übereinstimmten, jedoch häufig durch relative Methoden falsch dargestellt wurden.
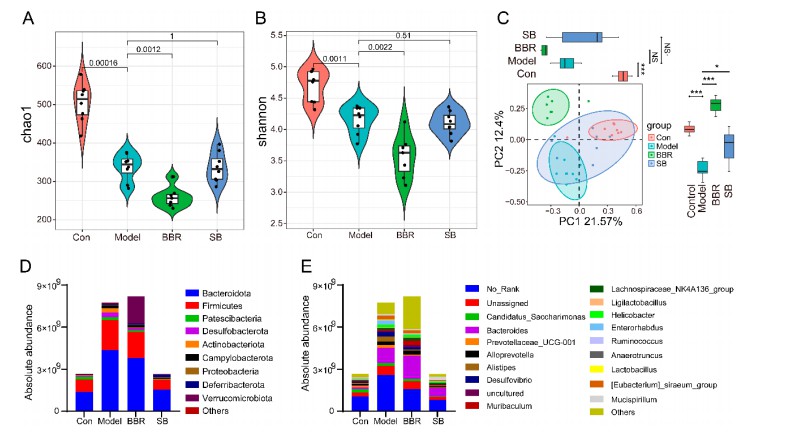 Abbildung. Absolute quantitative Analyse der Darmmikrobiota nach Behandlung mit Berberin (BBR) und Natriumbutyrat (SB) bei Mäusen mit DSS-induzierter Kolitis. Die Gemeinschaftsreichweite, -vielfalt und taxonomischen Profile wurden unter Verwendung von absoluten Abundanzdaten bewertet, was klarere Veränderungen als die relative Quantifizierung offenbarte.
Abbildung. Absolute quantitative Analyse der Darmmikrobiota nach Behandlung mit Berberin (BBR) und Natriumbutyrat (SB) bei Mäusen mit DSS-induzierter Kolitis. Die Gemeinschaftsreichweite, -vielfalt und taxonomischen Profile wurden unter Verwendung von absoluten Abundanzdaten bewertet, was klarere Veränderungen als die relative Quantifizierung offenbarte.
Schlussfolgerungen
Diese Studie zeigt, dass absolute quantitative metagenomische Sequenzierung bietet eine genauere Darstellung der Veränderungen in der mikrobielle Gemeinschaft als die relative Quantifizierung. Für Studien zu Arzneimitteln und Mikrobiomen, wie die anti-kolitis Wirkung von BBR, sind absolute Abundanzdaten:
- Vermeiden Sie falsche Korrelationen.
- Erfassen Sie echte mikrobielle Veränderungen,
- Bieten Sie stärkere translations- und klinische Relevanz.
Referenzen:
- Yang Y, Che Y, Liu L, Wang C, Yin X, Deng Y, Yang C, Zhang T. Schnelle absolute Quantifizierung von Pathogenen und ARGs durch Nanopore-Sequenzierung. Sci Total Environ2022 Feb 25;809:152190. doi: 10.1016/j.scitotenv.2021.152190. Epub 2021 Dez 7. PMID: 34890655.
- Barlow, J.T., Bogatyrev, S.R. & Ismagilov, R.F. Ein quantitatives Sequenzierungsrahmenwerk für absolute Abundanzmessungen von mukosalen und luminalen mikrobiellen Gemeinschaften. Nat Kommunizieren 11, 2590 (2020).


