
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von ProbenWas ist räumliche Transkriptomik?
Die räumliche Transkriptomik, ein innovativer Ansatz zur tiefen Analyse von RNA-seq-Daten in einer räumlichen Dimension, ermöglicht eine umfassende Darstellung der mRNA-Verteilung innerhalb jedes einzelnen Gewebeschnitts. Dies erlaubt anschließend die präzise Lokalisierung und differenzierte Identifizierung von aktiv exprimierten Funktionsgenen innerhalb eines bestimmten Gewebebereichs. Angesichts der Tatsache, dass Zellen, die die Grundeinheiten eines Organismus darstellen, ihre einzigartigen biologischen Funktionen in Zusammenarbeit mit ihrem räumlich spezifischen Mikroumfeld ausüben, wird die Beherrschung von Informationen zur räumlichen Positionierung von größter Bedeutung, wenn es darum geht, Mechanismen in der Zellbiologie, Onkologie und Entwicklungsbiologie zu untersuchen.
Dank der Fortschritte in der mikroskopischen Bildgebungstechnologie (einschließlich Superauflösung und Einzelmolekül-Bildgebung) sowie kontinuierlicher Verbesserungen der mehrfarbigen Fluoreszenz-in-situ-Hybridisierungstechniken hat unser Verständnis von Zell- und Gewebestruktur sowie deren Funktionen exponentiell zugenommen. Gleichzeitig hat die Entwicklung von Sequenzierungstechnologien präzise quantitative und qualitative Analysen der Genexpression in zuvor unerforschten Zellen oder Geweben ermöglicht. Die räumliche Transkriptomik, die mikroskopische Bildgebung und Sequenzierungstechnologien integriert, bewahrt so viele räumliche Informationen der Probe wie möglich, während sie Daten zur Genexpression beschafft. Dies bietet Wissenschaftlern revolutionäre Erkenntnisse mit tiefgreifenden Implikationen.
FFPE räumliche Transkriptomik-Sequenzierung
Mit dem kontinuierlichen Fortschritt und der Innovation in der Technologie der räumlichen Transkriptomik werden zahlreiche technische Herausforderungen allmählich überwunden. Kürzlich erzielte 10X Genomics einen bedeutenden Durchbruch, indem es die räumliche Transkriptomik anwendete, um erfolgreich die Einschränkungen von formalinfixierten, paraffineingebetteten (FFPE) Gewebeschnitten in der Analyse der räumlichen Genexpression zu adressieren. Diese Entwicklung bietet eine robuste Unterstützung für die weitere Erforschung räumlicher Informationen. Die Visium-Technologie zur räumlichen Genexpression kombiniert geschickt Histologie mit Hochdurchsatz-RNA-Sequenzierung und erweitert erheblich die Arten und den Umfang der Probenverarbeitung in der Einzelzell-Sequenzierungstechnologie. Dies bietet eine reichhaltigere Auswahl für die Forschung in Bereichen wie der Pathophysiologie und ermöglicht umfassende transkriptomische Analysen von über 18.000 Genen bei Menschen und Mäusen. Diese Technologie ermöglicht die eingehende Untersuchung der Genexpression von Signalwegen, die Analyse der Gewebeheterogenität und die Offenlegung von Zelltypen und -zuständen in verschiedenen gewebemorphologischen Hintergründen.
10x Genomics Visium Spatial Transcriptomics Sequenzierungsdienst bei CD Genomics
Der Spatial Transcriptomics Sequencing Service von CD Genomics ist eine bahnbrechende Technik, die die fortschrittliche 10x Visium-Plattform nutzt und eine präzise in-situ-transkriptionale Profilierung der gesamten Genexpression innerhalb von Geweben ermöglicht. Dieser Ansatz bietet detaillierte Einblicke nicht nur in die Ebenen der Genexpression, sondern lokalisiert auch genau, wo Gene im Gewebe exprimiert werden. Dieses Verfahren ermöglicht es uns, unterschiedliche Genexpressionen in verschiedenen funktionalen Regionen innerhalb von Geweben direkt zu beobachten und zu vergleichen. Dadurch bietet es ein leistungsstarkes Werkzeug für die eingehende Erforschung von Gewebefunktionen und Krankheitsmechanismen und verstärkt somit unsere Fähigkeit, die Mechanismen von Gesundheit und Krankheit zu ergründen.
 10x Genomics
10x Genomics
10x Genomics Visium räumliche Transkriptomik Sequenzierungsprinzipien
Der Schlüssel zur Technologie hinter räumlichen Genexpressionslösungen liegt im Folienabschnitt. Als Grundlage für den Bibliotheksaufbau dient jede Folie, die mit vier präzisen Erfassungsbereichen gemustert ist, die jeweils 6,5 x 6,5 Millimeter messen und in einem quadratischen Rahmen dargestellt sind. In jedem Erfassungsbereich befinden sich 5000 barcode-gekennzeichnete Punkte, die einen Durchmesser von 55 Mikrometern haben und einen Abstand von 100 Mikrometern zwischen benachbarten Punkten aufweisen. Jeder Punkt trägt eine einzigartige Barcode-Sequenz – einen räumlichen Barcode – der verwendet wird, um die einzelnen Punkte zu identifizieren und zu unterscheiden. Darüber hinaus hat jede Nukleinsäuresonde innerhalb eines Punktes ein exklusives einzigartiges Tag, um verschiedene Transkripte innerhalb einer einzelnen Zelle zu unterscheiden und PCR-Duplikate zu beseitigen, wodurch eine absolute Quantifizierung erreicht wird.
Nach der Freisetzung von RNA aus den Zellen im Gewebeschnitt wandern sie zu den zuvor genannten Stellen und werden mit entsprechenden Barcode-Sequenzen versehen. Anschließend durchläuft die RNA die Prozesse der Bibliothekskonstruktion und Sequenzierung. Durch die Nutzung der Barcode-Informationen aus den Proben-Daten können wir die Daten genau ihrem relevanten Standort zuordnen und die Ursprünge der Daten aus spezifischen räumlichen Koordinaten festlegen. Letztendlich ermöglicht dieser Prozess die Visualisierung der räumlichen Genexpression.
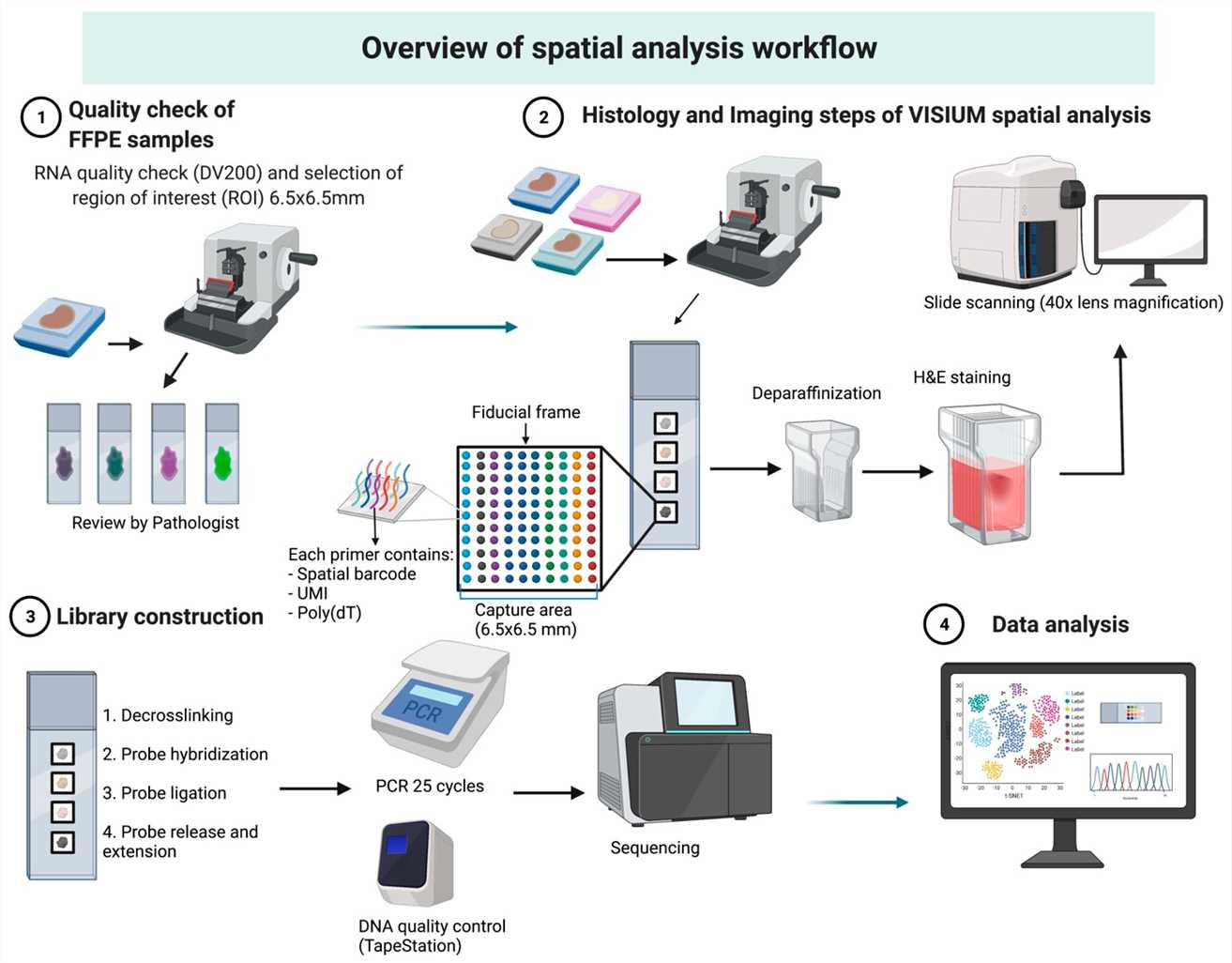 Übersicht über den Visium-Raumanalysen-Workflow. (Dmitrii Shek et al.) Methoden Protokolle. 2023)
Übersicht über den Visium-Raumanalysen-Workflow. (Dmitrii Shek et al.) Methoden Protokolle. 2023)
Experimentelle Schritte der räumlichen Transkriptomik-Sequenzierung
Präzises Schneiden von frisch gefrorenen Gewebeproben wird durchgeführt, gefolgt von der Visualisierung mit fortschrittlichen Bildgebungstechniken.
Die Gewebeschnitte werden auf einem speziell gestalteten Objektträger platziert, der mit RNA-spezifischen Fangsonden beschichtet ist. Nach strengen Fixierungs- und Permeabilisierungsschritten wird sichergestellt, dass die mRNA aus den Zellen vollständig freigesetzt und spezifisch an die Fangsonden gebunden wird, was zu einer genauen Erfassung von Informationen über die Genexpression führt.
Erfasste RNA dient als Vorlage für die präzise cDNA-Synthese und die Vorbereitung der Sequenzierungsbibliothek, wodurch die Genauigkeit und Zuverlässigkeit der nachfolgenden Analyse gewährleistet wird.
Die vorbereiteten Sequenzierungsbibliotheken durchlaufen eine effiziente Sequenzierung für eine umfassende Erfassung von Genexpressionsinformationen.
Durch die Analyse von Datenvisualisierungen bestimmen wir präzise die exprimierten Gene, quantifizieren sie und interpretieren darüber hinaus ihre räumlichen Positionsinformationen.
Workflow für den Sequenzierungsdienst der räumlichen Transkriptomik
Visium Spatial Gene Expression für frisches gefrorenes Gewebe Service-Workflow
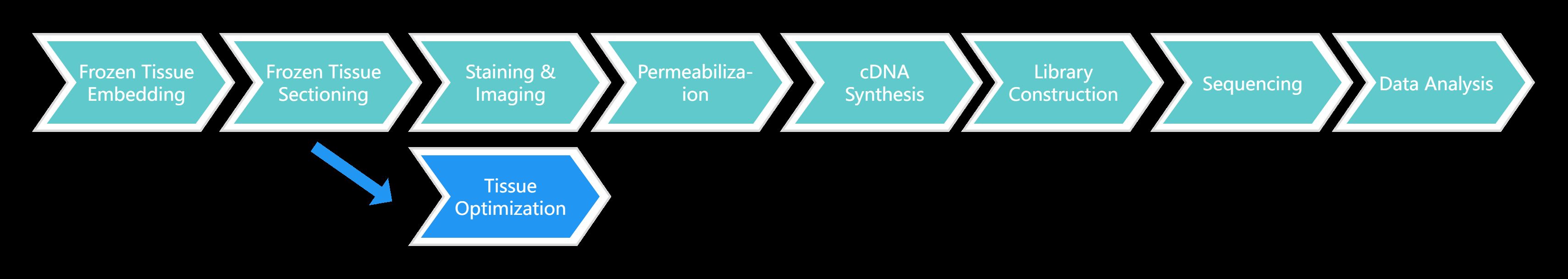
Visium Spatial Gene Expression für FFPE-Service-Workflow

Dienstspezifikation
| Beispielanforderungen Frisches Gewebe: 6,5 mm³ FFPE: 6,5 mm³; DV200 > 50% Artenbereich: Mensch, Maus, Ratte. Für andere Arten konsultieren Sie bitte. |
|
 |
Sequenzierung Sequenzierungsplattform: Illumina NovaSeq 6000 Sequenzierungsmuster: PE150 Sequenzierung Datenvolumen: ≥50k Lese-Paare pro Spot. |
 |
Bioinformatikanalyse Wir bieten maßgeschneiderte bioinformatische Analysen an, einschließlich: Rohsequenzdaten Qualitätsbewertung und Filterung von Sequenzierungsdaten Stichprobenqualitätskontrolle Datenanpassung Datenstandardisierung Punktclustering Spot-Unterpopulationanalyse Markeranalyse Zelltypidentifikation Anatomische Regionsannotation Zellanalyse der Kommunikation Inter-Sample-Differenzialanalyse Differenzanalyse im Inter-Annotation-Bereich |
Probenvorbereitung
Es ist wichtig, eine entscheidende Vorsichtsmaßnahme im Prozess der Probenvorbereitung zu erwähnen, insbesondere für Gewebeproben. Es wird empfohlen, das Schnelleinfrieren in flüssigem Stickstoff zu vermeiden, da die schnelle Abkühlung potenziell Schäden an Zellstrukturen verursachen kann. Stattdessen wird die Verwendung von in OTC eingebettetem Pentan für das schnelle Einfrieren von Gewebeproben offiziell von 10X Genomics empfohlen. In der praktischen Anwendung stoßen viele Forscher häufig auf Schwierigkeiten bei der Vorbereitung der Suspension während Einzelzell-Experimenten. Trotz wiederholter Versuche bei der Verdauungsverarbeitung ist es nach wie vor mühsam, eine Einzelzell-Suspension zu erhalten, die den Anforderungen für die Maschinenbenutzung entspricht. Selbst wenn man widerwillig mit dem Maschinenbetrieb fortfährt, könnten Risiken für suboptimale Zellbedingungen, unzureichende Zellaufnahme und die Komplikation von mehrzelligen Verunreinigungen bestehen. Im Vergleich dazu ist der Arbeitsablauf von Experimenten zur räumlichen Transkriptomik etwas gestrafft. Unter der Anleitung erfahrener Unternehmen können nach einer angemessenen Einfrierung von Gewebeproben die nachfolgenden Schritte der Gewebeaufklärung, Färbung, Fixierung, Klarstellung, Bibliothekskonstruktion, Sequenzierung und Analyse usw. an professionelle Unternehmen übertragen werden. Dies könnte den Forschern eine beträchtliche Menge an Zeit sparen.
Servicevorteile
Überlegene Qualität beim SchneidenMit umfangreicher Erfahrung im Schneiden und Montieren haben wir optimierte Lösungen für verschiedene Gewebe entwickelt.
Automatisierte AnalyseUnser gut etabliertes Analyseverfahren bietet eine schnelle und präzise Interpretation von räumlichen Transkriptomdaten.
Standardisierte QualitätskontrolleUnser Reichtum an praktischer Erfahrung hat zur Entwicklung eines standardisierten Qualitätskontrollsystems geführt.
Professionelles TeamUnser renommiertes Technikteam verfügt über jahrelange Erfahrung in der Projektplanungsdesign, Laborbetrieb und Nachverkaufsanalyse.
Vollservice-AnsatzWir bieten ein umfassendes Dienstleistungsangebot, das Gewebe-Kryokonservierung und Einbettung, Montage, Schneiden, Klärung, Bibliothekskonstruktion und Sequenzierung sowie Datenanalyse umfasst.
Demo-Ergebnisse
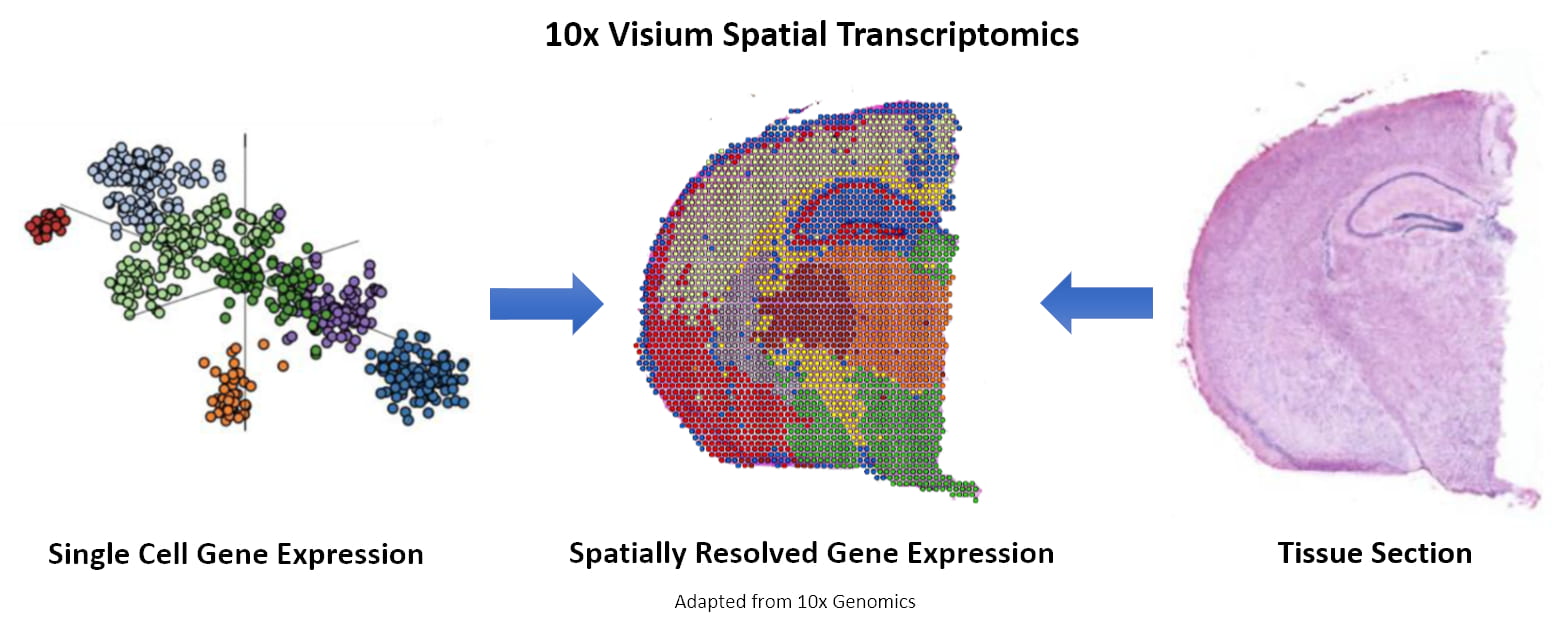 Abbildung 1. Punktclusterung und Bildintegration. (Quelle: 10x Genomics)
Abbildung 1. Punktclusterung und Bildintegration. (Quelle: 10x Genomics)
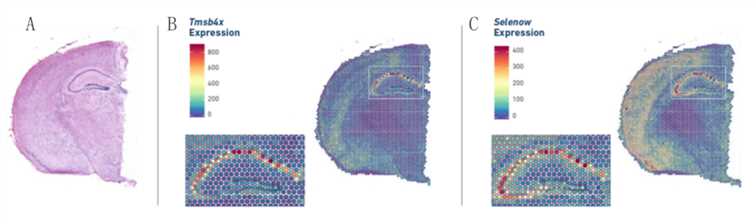 Abbildung 2. Genexpressionsmuster in räumlicher Auflösung. (Quelle: 10x Genomics)
Abbildung 2. Genexpressionsmuster in räumlicher Auflösung. (Quelle: 10x Genomics)
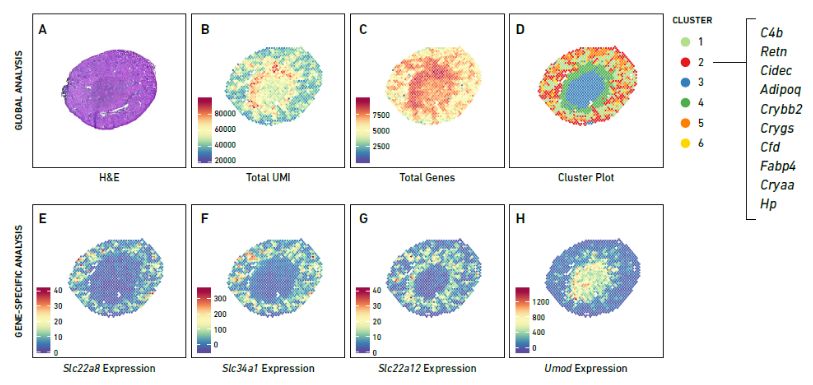 Abbildung 3. mRNA-Expression und Clusterkarte des räumlichen Transkriptoms. (Quelle: 10x Genomics)
Abbildung 3. mRNA-Expression und Clusterkarte des räumlichen Transkriptoms. (Quelle: 10x Genomics)
10x Spatial Transcriptomik Seq FAQs
10x räumliche Transkriptom-Sequenzierungs-Fallstudien
Räumliche Kartierung zeigt menschliche Adipozyten-Subpopulationen mit unterschiedlichen Empfindlichkeiten.
Zeitschrift: Zellstoffwechsel
Impact-Faktor: 27,287
Veröffentlicht: 7. September 2021
Hintergrund
Weißes Fettgewebe (WAT) reguliert den Energiehaushalt durch Fettspeicherung und die Sekretion von Faktoren. Seine Plastizität umfasst verschiedene Zelltypen, wobei eine gesunde WAT-Erweiterung Gefäßbildung, Adipozytenbildung und Immunantworten benötigt, während ungesundes WAT Hypertrophie und chronische Entzündungen zeigt, die zu Stoffwechselerkrankungen führen. Die räumliche Transkriptomik in Kombination mit Einzelzell-RNA-Sequenzierung hat drei verschiedene Adipozytenpopulationen im menschlichen WAT identifiziert, die jeweils unterschiedliche hormonelle Reaktionen aufweisen und die Bedeutung der Adipozyten-Heterogenität in der hormonellen Sensitivität hervorheben.
Materialien & Methoden
Probenvorbereitung
- Menschliche Probanden
- Subkutane Fettgewebeproben
Sequenzierung
- 10x räumliche Transkriptom-Sequenzierung
- Illumina NovaSeq6000 Plattform
- Sequenzdatenverarbeitung
- Bildbasierte Bewertungen der Fettzellgröße
- Analysen von Expressionsprofilen und -wegen
- Analysen von Marker-Genen der Adipozyten-Subtypen
Ergebnisse
Mit der 10x Genomics Visium Spatial Gene Expression-Plattform analysierten die Autoren subkutanes abdominales WAT von zehn Individuen. Sie identifizierten 20 verschiedene Zellklassen, darunter Immun-, Gefäß- und Vorläuferzellen. Die Kombination aus räumlicher Transkriptomik und Einzelzell-RNA-Sequenzierung offenbarte drei reife Adipozytenklassen mit einzigartigen Gen-Signaturen. Diese Ergebnisse unterstreichen die zelluläre Heterogenität und räumliche Organisation innerhalb des WAT und erweitern das Verständnis seiner komplexen Struktur und Funktion.
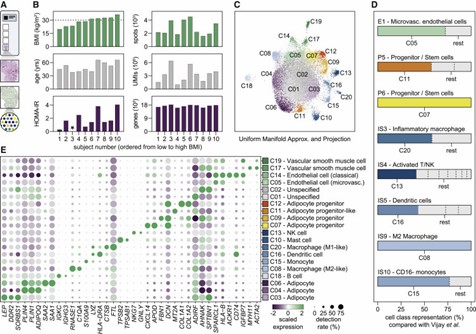 Abbildung 1. Integrative Analysen von räumlichen und Einzelzell-Transkriptomdaten identifizieren 18 verschiedene Zellklassen im menschlichen WAT.
Abbildung 1. Integrative Analysen von räumlichen und Einzelzell-Transkriptomdaten identifizieren 18 verschiedene Zellklassen im menschlichen WAT.
Die Autoren analysierten systematisch die zelluläre Organisation des weißen Fettgewebes (WAT) und entdeckten, dass Zellen sowohl homotypische als auch heterotypische Cluster bilden. Immunzellen, Adipozytenvorläufer und Gefäßzellen zeigten unterschiedliche Grade der homotypischen Clusterbildung. Darüber hinaus offenbarte die Studie räumliche Beziehungen zwischen verschiedenen Zellklassen. Diese Ergebnisse heben die komplexen und räumlich definierten Zell-Zell-Beziehungen innerhalb des WAT hervor.
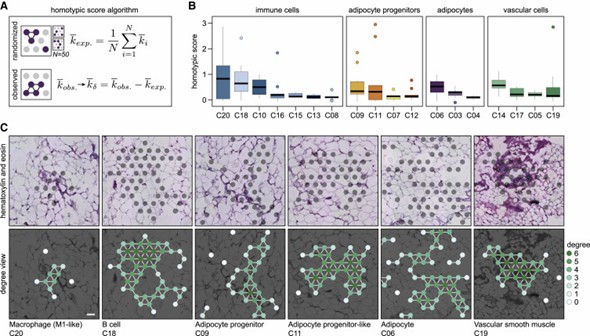 Abbildung 2. WAT-Residentenzellen zeigen große Variationen in der Neigung, homotypische Zellcluster zu bilden.
Abbildung 2. WAT-Residentenzellen zeigen große Variationen in der Neigung, homotypische Zellcluster zu bilden.
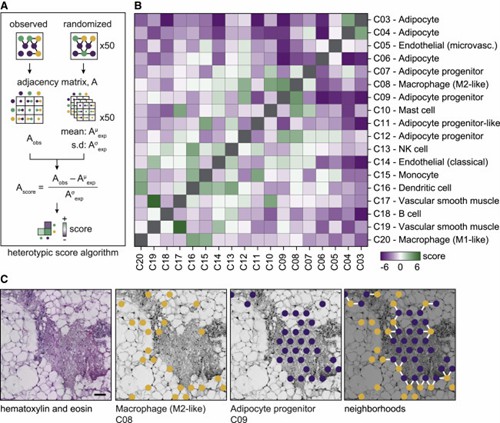 Abbildung 3. Bestimmte Zellpopulationen zeigen heterotypische Cluster im WAT.
Abbildung 3. Bestimmte Zellpopulationen zeigen heterotypische Cluster im WAT.
Fazit
Die Autoren verwendeten hochauflösende räumliche Transkriptomik, um menschliches weißes Fettgewebe (WAT) zu kartieren, und identifizierten 18 verschiedene Zellklassen, darunter Adipozytenvorläufer, Immun-, Gefäß- und reife Fettzellen. Sie entwickelten Algorithmen, um homotypische und heterotypische Zellcluster zu entdecken, und zeigten, dass WAT strukturell organisierter ist als bisher angenommen. Bemerkenswert ist, dass nur eine der drei identifizierten reifen Adipozytenpopulationen signifikant auf hormonelle Stimuli reagierte. Diese Arbeit erweitert unser Verständnis der zellulären Architektur von WAT und deren Einfluss auf die hormonelle Sensitivität und bietet eine wertvolle Ressource für weitere Forschungen.
Referenz
- Bäckdahl J, Franzén L, Massier L, et al. Räumliche Kartierung zeigt menschliche Adipozyten-Subpopulationen mit unterschiedlichen Empfindlichkeiten. Zellstoffwechsel, 2021, 33(9): 1869-1882. e6.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die HLA-Klasse-I-Immunopeptidome der AAV-Kapsidproteine
Zeitschrift: Frontiers in Immunologie
Jahr: 2023
Isolation und Charakterisierung neuer menschlicher Trägerpeptide aus zwei wichtigen Impfstoff-Immunogenen
Zeitschrift: Impfstoff
Jahr: 2020
Änderung des Gewichts, des BMI und der Körperzusammensetzung in einer bevölkerungsbasierten Intervention im Vergleich zu einer genetisch basierten Intervention: Die NOW-Studie
Zeitschrift: Fettleibigkeit
Jahr: 2020
Sarecyclin hemmt die Proteintranslation im Cutibacterium acnes 70S-Ribosom durch einen Zwei-Stellen-Mechanismus.
Zeitschrift: Nucleic Acids Research
Jahr: 2023
Identifizierung eines Darmkommensalen, der die blutdrucksenkende Wirkung von Ester-Angiotensin-Converting-Enzym-Hemmern beeinträchtigt.
Zeitschrift: Hypertonie
Jahr: 2022
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Zeitschrift: Zellen
Jahr: 2021
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


