
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Genpanel-Sequenzierungsdienst
Was sind WES, WGS und Panel-Sequenzierung?
Whole Genome Sequencing (WGS) und Whole Exome Sequencing (WES) sind abgekürzte Begriffe für die Sequenzierung des gesamten Genoms und des gesamten Exoms, jeweils. Im Kontext von Next-Generation Sequencing (NGS)Der Begriff "Panel" steht für ein "Genpaket" oder eine Gruppe gezielter Gene.
Merkmale von WGS, WES und Panel-Sequenzierung
WGS, WES und Panel weisen jeweils unterschiedliche Merkmale im Bereich der Genomik auf.
Um dies zu veranschaulichen, umfasst die gesamte Genomsequenz etwa 3 Gigabasen (Gb). Wenn die Sequenzierungsdaten 10 Gb betragen, kann das gesamte Genom ungefähr 3 Mal abgedeckt werden, was als 3X Sequierungstiefe bezeichnet wird. Im Gegensatz dazu können die kodierenden Exons, die nur 1% des Genoms oder etwa 30 Megabasen (Mb) groß sind, mit 10 Gb Sequenzierungsdaten Sequierungstiefen von über 100X erreichen. Dies ist noch ausgeprägter bei Panels, die auf spezifische Gene abzielen, wo 1 Gb Daten Tiefen von etwa 1000X erreichen kann. Ein reduziertes Datenvolumen korreliert mit niedrigeren Kosten, aber ein wesentlicher Vorteil von Panels liegt in ihrer hohen Sequierungstiefe.
Was sind die Vorteile einer hohen Sequenzierungstiefe??
Betrachten Sie ein Wildtyp-Locus mit dem GG-Genotyp. Wenn eine heterozygote Mutation auftritt, wird der Genotyp zu GT. Sequenzierungsdaten zeigen gleichzeitig Sequenzen, die sowohl G als auch T enthalten, ungefähr im Verhältnis 1:1. In genetischen Tests werden Sequenzierungsdaten verwendet, um den tatsächlichen Genotyp abzuleiten. Um mit Sicherheit auf eine heterozygote Mutation an einem bestimmten Locus zu schließen und Störungen durch Sequenzierungsfehler zu vermeiden, ist eine bestimmte Menge an Varianten-Sequenzen erforderlich, was eine minimale Sequenzierungstiefe, wie z.B. 20X, notwendig macht.
Aufgrund systematischer Verzerrungen in den Sequenzierungs- und Analyseprozessen könnte die Verteilung von Sequenzen mit G und T kein perfektes 1:1-Verhältnis aufweisen. Darüber hinaus kann die Sequenzierungstiefe an verschiedenen Loci variieren, beeinflusst von Faktoren wie dem genomischen GC-Gehalt und der Spezifität der Sonden während der Erfassung. Eine höhere Sequenzierungstiefe mildert diese Probleme und verringert die Wahrscheinlichkeit, dass Varianteninformationen in problematischen Regionen fehlen.
Darüber hinaus erweist sich eine substanzielle Sequierungstiefe als vorteilhaft bei der Identifizierung von Mosaikvariationen. In Fällen, in denen Variationen während der Befruchtungsphase auftreten, werden die Varianten-Sequenzen, unabhängig von der Anzahl der sequenzierten Zellen, jeweils etwa die Hälfte ausmachen. Im Gegensatz dazu können Variationen, die in intermediären Entwicklungsstadien auftreten, Mosaikmuster aufweisen. Die Fähigkeit, niedrigere Anteile von Mosaizismus zu erkennen, erfordert eine erhöhte Sequierungstiefe, wodurch Gen-Panels besonders gut für diese Aufgabe geeignet sind.
Zusammenfassung der Eigenschaften von drei Sequenzierungstechnologien
Gen-Panels zeigen die Fähigkeit, eine erhöhte Sequenzierungstiefe zu erreichen, was eine vorteilhafte Position in der genetischen Analyse verleiht. Darüber hinaus bieten sie erhebliche Anpassungsflexibilität, die die Einbeziehung pathogener Regionen innerhalb nicht-kodierender Bereiche erleichtert.
WESstrategisch positioniert zwischen WGS und die Panel-Sequenzierung schafft ein harmonisches Gleichgewicht, indem sie einen erweiterten Sequenzierungsumfang zu allgemein akzeptablen Kosten bietet. Diese Technologie ermöglicht es Forschern, eine umfassende Sequenzierungsabdeckung zu erreichen, während eine bemerkenswerte Sequenzierungstiefe aufrechterhalten wird.
WGS zeichnet sich durch seinen unvergleichlichen Sequenzierungsumfang aus, der das gesamte Genom umfasst. Um jedoch Basenpaarvariationen zu erkennen, ist ein umfangreicher Datensatz von etwa 100 Gigabasen (Gb) erforderlich. Die aktuellen Herausforderungen in Bezug auf die Machbarkeit und Erschwinglichkeit des Erwerbs solch umfangreicher Daten unterstreichen die Überlegungen für eine breite Anwendung.
Wie man Panel, WES oder WGS auswählt
Die laufende Debatte im Bereich der genetischen Tests dreht sich um die Auswahl von Panel, WES oder WGS. Jede Methode bietet besondere Vorteile, was einen durchdachten Entscheidungsprozess erforderlich macht.
In Fällen, in denen die Testziele klar definiert sind, sollte die anfängliche Präferenz auf die Panel-Sequenzierung gerichtet sein, um eine erhöhte Sensitivität innerhalb des festgelegten Erkennungsbereichs zu gewährleisten. Umgekehrt sollten bei weniger klaren Testzielen die Überlegungen in Richtung WES oder WGS um ein breiteres Spektrum pathogener Faktoren aufzudecken. WGS umfasst die Sequenzierung jedes Basenpaars des Genoms, während WES und Panel-Sequenzierung gezielte Sequenzierungstechniken verwenden.
Letztendlich hängt der Entscheidungsprozess von der Klarheit der Testziele ab. Die Wahl der Panel-Sequenzierung gewährleistet Präzision bei der Erkennung bekannter Ziele, während WES oder WGS bevorzugt werden können, wenn das Ziel darin besteht, eine breitere Palette potenzieller pathogener Faktoren zu untersuchen, insbesondere in Szenarien, in denen die Testziele an Spezifität mangeln.
Unsere Panel-Sequenzierungsdienste
CD Genomics bietet präzise und kosteneffektive vordesignte sowie maßgeschneiderte NGS-Panel-Dienste an, die das Erfassen von DNA-Fragmenten aus mehreren relevanten Genzielregionen unter Verwendung spezifischer Genfangsonden umfassen. Anschließend werden die erfassten DNA-Sequenzen in den Zielregionen bestimmt mithilfe von NGS Technologie, die die Identifizierung von Zielgenen und Mutationsstellen ermöglicht.
RNA-Sequenzierungs-Panel
Die gezielte RNA-Sequenzierung ist die optimale Wahl für das Studium der Genexpression und Umstellungen, selbst bei Proben von schlechter Qualität wie FFPE und cfRNA. Die Proben-Technologie, die alle exonspezifischen Regionen der interessierenden Gene anvisiert, bietet eine hohe Abdeckung und ermöglicht eine umfassende Analyse der Genexpression, einschließlich der Subtypanalyse der totalen RNA-Sequenzierung.
Exom-Sequenzierungs-Panel
Exom-Sequenzierungspanels, die als weit verbreitete Methode in der Krankheitsforschung eingesetzt werden, können pathogenetische Genvariationen in Zielgenen über das gesamte Genom schnell und effektiv nachweisen. Dieser Ansatz verbessert die Datenabdeckungstiefe und reduziert effektiv die Analysezeit und die Sequenzierungskosten.
Gezielte Methylierungssequenzierungs-Panels
CD Genomics hat eine Proben-Technologie basierend auf Bisulfit-Konversion eingeführt, die eine Methylierungsanalyse verschiedener Probenarten wie gDNA und cfDNA ermöglicht. Diese Methode analysiert den Methylierungsstatus von Zielgenen genau und liefert umfassende Methylierungsergebnisse für die Forschung. Anschließend bietet CD Genomics auch umfassende bioinformatische Analyse-Dienste für Methylierungsdaten an.
Warum Sie sich für unsere maßgeschneiderten NGS-Sequenzierungs-Panels entscheiden sollten
- Genauigkeit: Basierend auf der Tiefensequenzierungstechnologie übersteigt die Genauigkeit der Ergebnisse 99 %.
- Spezifische Erfassung: Zeigt hervorragende Spezifität, die eine chromosomspezifische Erfassung in polyploiden Organismen ermöglicht.
- Flexible Anpassung: Hochgradig anpassungsfähig, in der Lage, gleichzeitig die Gensequenzdetektion und die gezielte Mutationsdetektion in einer einzigen Pipeline durchzuführen.
- Hohe Erkennungseffizienz: Ermöglicht die parallele Sequenzierung von Hunderttausenden bis Millionen von DNA-Molekülen in einem einzigen Durchlauf, was die gleichzeitige Erkennung mehrerer Krankheiten, zahlreicher Gene und zehntausender Mutationsstellen erleichtert. Dies gewährleistet schnelle, effiziente, zuverlässige Ergebnisse mit hoher Durchsatzrate und Kosteneffektivität.
Anwendung der Genpanel-Sequenzierung
- Genetische ForschungEntdecken Sie neuartige Varianten, untersuchen Sie Genfunktionen und verstehen Sie biologische Prozesse sowie Krankheitsmechanismen.
- KrankheitsassoziationsstudienIdentifizieren Sie Gene, die mit genetischen Erkrankungen oder komplexen Krankheiten verbunden sind, und untersuchen Sie genetische Variationen in verschiedenen Populationen.
- KrebsforschungProfilieren Sie krebsbezogene Mutationen, klassifizieren Sie Krebsuntertypen und untersuchen Sie die Tumorentstehung.
- PharmakogenomikAnalysieren Sie genetische Faktoren, die die Arzneimittelreaktionen beeinflussen, und identifizieren Sie Marker für Arzneimitteltoxizität.
- Funktionale ValidierungValidieren Sie Genfunktionen und untersuchen Sie Interaktionen innerhalb zellulärer Signalwege.
- Biomarker-EntdeckungIdentifizieren Sie diagnostische und prognostische genetische Marker zur Krankheitsdetektion und -progression.
- Untersuchung seltener KrankheitenFinde Gene, die mit seltenen Erkrankungen assoziiert sind, und untersuche genetische Vererbungsmuster.
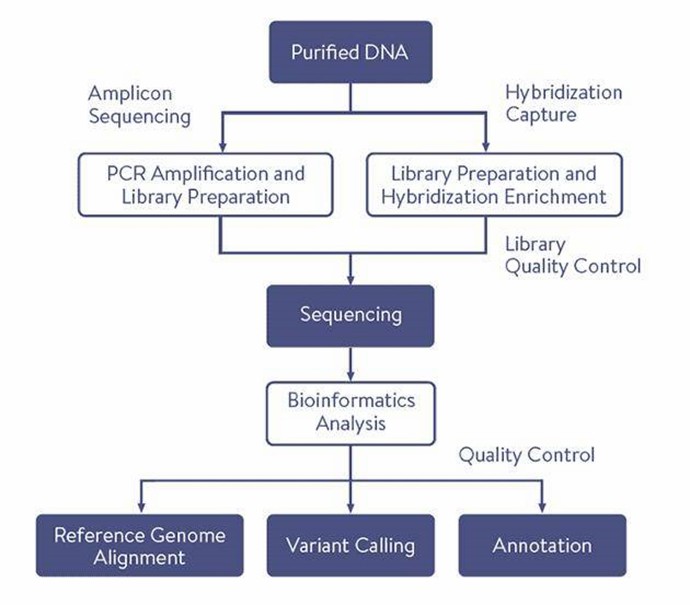
Gen-Pane-Sequenzierungs-Workflow
Der Gene Panel Sequencing Service von CD Genomics bietet eine präzise und effiziente Lösung für gezielte genetische Forschung. Der Service umfasst das Design maßgeschneiderter Gen-Panels, Hochdurchsatz-Sequenzierung und detaillierte Datenanalyse, um wichtige genetische Varianten zu identifizieren, die für Ihre Forschungsziele relevant sind.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
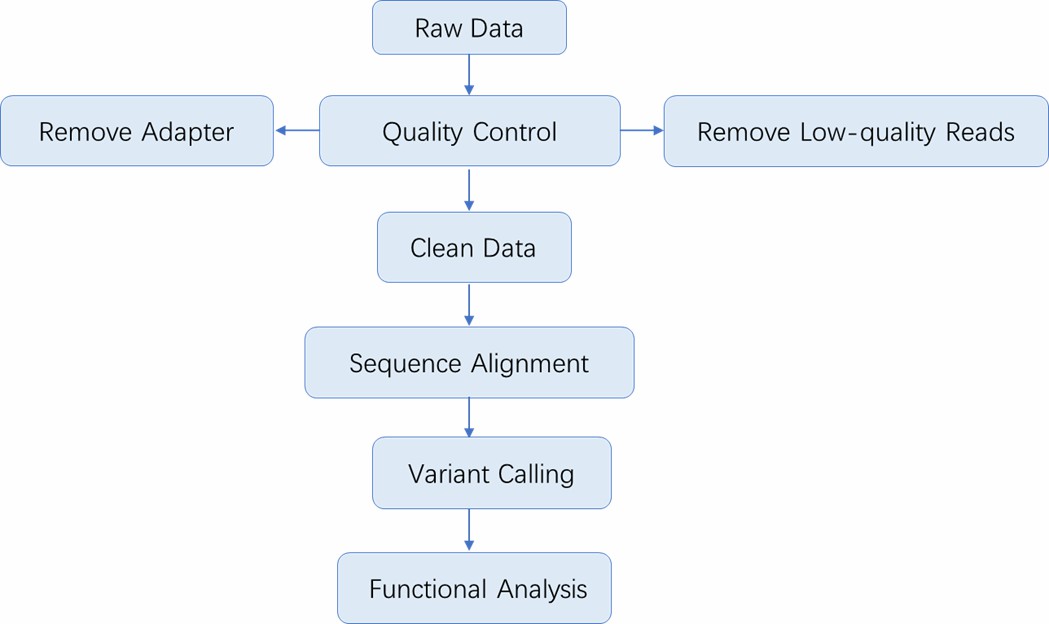
Bioinformatische Analyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Genpanel-Sequenzierung für Ihre Schreibanpassung
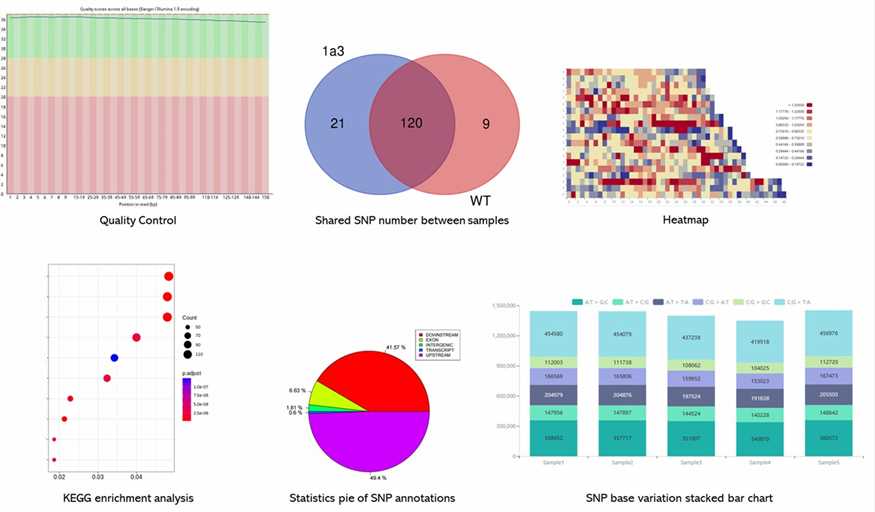
Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
Gene-Panel-Sequenzierungs-FAQs
1. Was bedeuten "kleines Panel" und "großes Panel" bei der Auswahl von genetischen Testoptionen?
Im Kontext von NGS„kleines Panel“ und „großes Panel“ beziehen sich auf unterschiedliche Bereiche basierend auf der Anzahl der analysierten Gene.
Kleines Panel: Dies umfasst typischerweise eine Gruppe von Genen, die von einem Dutzend bis zu einigen Dutzend reichen. Diese Panels konzentrieren sich in der Regel auf wichtige Treiber-Gene, die mit genehmigten oder klinisch relevanten zielgerichteten Therapien für spezifische Krebsarten in Verbindung stehen. Sie können auch eine Auswahl von Tumorsuppressorgenen enthalten, die erhebliches Forschungsinteresse geweckt haben.
Große Panels: Diese Panels umfassen Hunderte bis Tausende von Genen. Sie decken nicht nur Treiber-Gene ab, die für gezielte Therapien relevant sind, sondern beinhalten auch ein breites Spektrum von Genen, die mit Krebs assoziiert sind, soweit es die aktuelle Forschung und technologische Möglichkeiten zulassen. Die Ergebnisse aus großen Panels gehen über die Identifizierung gezielter Therapien für spezifische Krebserkrankungen hinaus und schließen auch krebsübergreifende Arzneimitteloptionen, immuntherapiebezogene Marker wie Tumormutationslast (TMB) und Mikrosatelliteninstabilität (MSI) sowie potenziell erbliche Krebsgene ein.
2. Wie unterscheidet sich die Genpanel-Sequenzierung von Whole-Genome-Sequenzierung?
Die Genpanel-Sequenzierung konzentriert sich auf vordefinierte Sätze von Genen oder genomischen Regionen und liefert hochauflösende Daten für diese gezielten Bereiche. Im Gegensatz dazu umfasst die Whole-Genome-Sequenzierung die Untersuchung des gesamten Genoms, einschließlich sowohl kodierender als auch nicht-kodierender Regionen. Dieser Ansatz bietet eine umfassende Übersicht über alle genetischen Variationen innerhalb eines Organismus.
3. Wie wähle ich das richtige Genpanel für meine Forschung aus?
Berücksichtigen Sie die spezifischen Gene oder Signalwege von Interesse, die Forschungsziele und vorhandenes Wissen über die beteiligten genetischen Faktoren. Konsultieren Sie einen Genom-Spezialisten oder überprüfen Sie verfügbare Panels, um sicherzustellen, dass sie mit Ihren Forschungszielen übereinstimmen.
Gene-Panel-Sequenzierungs-Fallstudien
Identifizierung von Mutationsgenen als prognostische Biomarker bei multiplem Myelom durch Exom-Sequenzierung von Gen-Panels und Transkriptomanalyse in der chinesischen Bevölkerung.
Journal: Computerbiologie und Medizin
Impactfaktor: 7,7
Veröffentlicht: September 2023
Hintergrund
Das multiple Myelom (MM) ist eine verbreitete Blutkrebserkrankung, die hauptsächlich ältere Erwachsene betrifft und bei Kindern in China seltener vorkommt. Es zeigt regionale Unterschiede in der Inzidenz und hat eine fünfjährige Überlebensrate von unter 40 %. Effektive Behandlungen fehlen derzeit. NGS ist wertvoll für das Verständnis von MM, indem kritische Mutationen identifiziert werden. Diese Studie verwendet Exom-Sequenzierung bei 50 chinesischen MM-Patienten, um bedeutende genetische Veränderungen und potenzielle neue Ziele für die Behandlung aufzudecken.
Materialien & Methoden
Probenvorbereitung
- Klinische Proben
- Knochenmarkproben
Sequenzierung
- Gezieltes Genpanel
- Whole-Exom-Sequenzierung
- Illumina MiSeq
- DEG-Identifizierung
- GO- und KEGG-Anreicherungsanalyse
- Überlebensanalyse
- Statistische Analyse
Ergebnisse
Die Exom-Sequenzierung von 50 MM-Patienten, die 400 Gene anvisierte, offenbarte verschiedene Mutationsarten, wobei nicht-synonyme Einzel-Nukleotid-Varianten (SNVs) vorherrschend waren. Die häufigsten Mutationen waren C > T-Substitutionen, und jede Probe wies median 576 Mutationen auf. Die zehn am häufigsten mutierten Gene waren MUC16, MUC4, TTN, AHNAK2, MUC17, OBSCN, OR4C3, MUC2, MKI67 und PRUNE2, die alle eine Mutationshäufigkeit von 100 % zeigten.
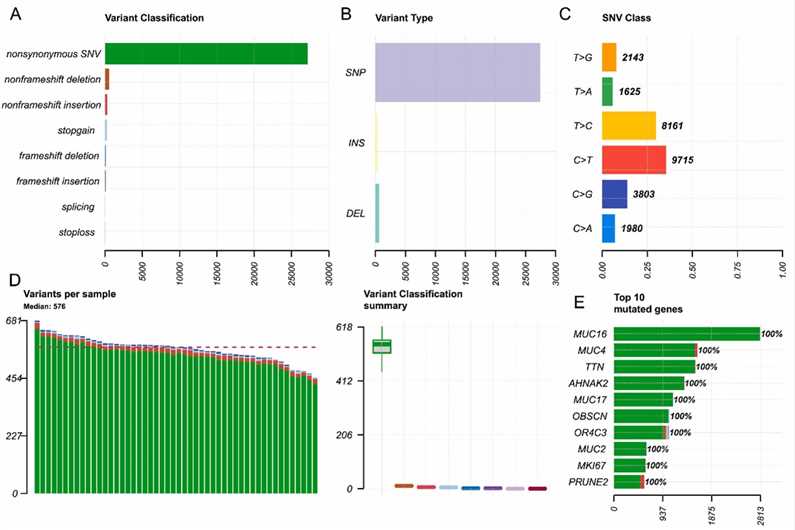 Abb. 1. Übersicht über den Mutationsstatus der 50 Patienten mit MM.
Abb. 1. Übersicht über den Mutationsstatus der 50 Patienten mit MM.
Die Studie analysierte die Mutationslandschaft von 50 MM-Patienten mithilfe eines 400-Gen-Panels. Es wurden Mutationen in 337 Genen identifiziert, wobei 48 Gene eine Mutationshäufigkeit von 100 % und 31 mehr als 90 % aufwiesen. Zu den wichtigsten mutierten Genen gehören CACNA1I, ID3 und EGFR. Die funktionelle Anreicherung zeigte, dass die mutierten Gene mit DNA-Modifikationen, Funktionen der extrazellulären Matrix und mehreren wichtigen Signalwegen, einschließlich PI3K-Akt und Notch, assoziiert waren.
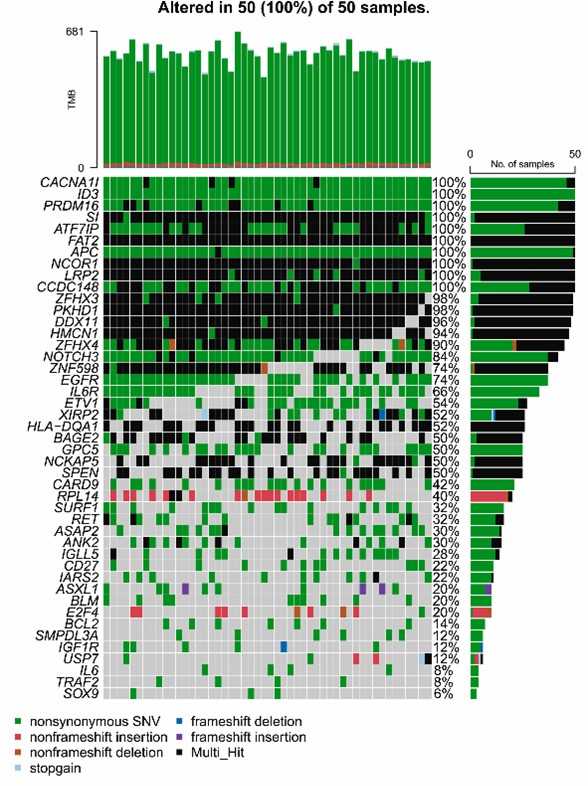 Abb. 2. Die 45 häufig mutierten Gene von 50 Patienten wurden mit OncoPrinter visualisiert.
Abb. 2. Die 45 häufig mutierten Gene von 50 Patienten wurden mit OncoPrinter visualisiert.
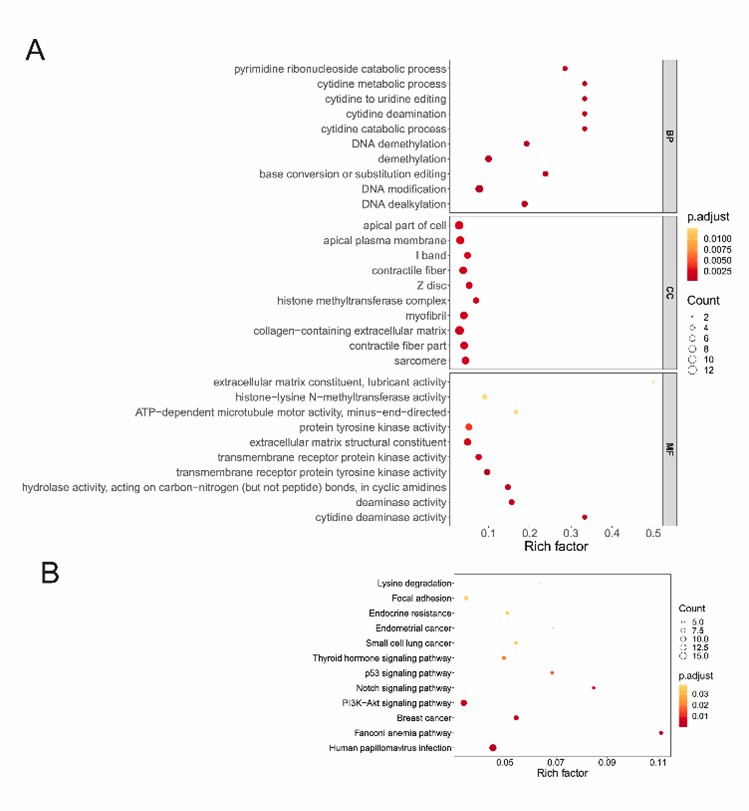 Abb. 3. Funktionsanreicherungsanalyse von Mutantgenen in MM.
Abb. 3. Funktionsanreicherungsanalyse von Mutantgenen in MM.
Die Analyse der Genexpression aus dem GSE6477-Datensatz identifizierte 1247 differentielle exprimierte Gene (DEGs), darunter 660 herunterregulierte und 587 hochregulierte. Zu den bemerkenswerten DEGs gehören RNASE2 und RPS19. Die Analyse der gemeinsamen Gene zwischen mutierten Genen und DEGs hob 33 Gene hervor, darunter ID3 und MYC, die in Wegen angereichert waren, die mit Zellproliferation, myeloider Differenzierung und viraler Karzinogenese in Zusammenhang stehen.
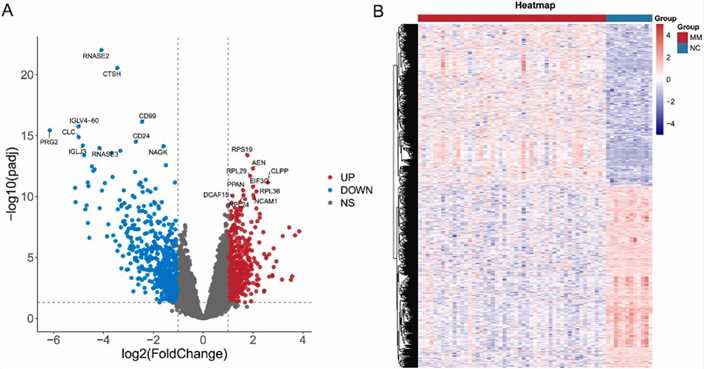 Abb. 4. Identifizierung von DEGs in den GSE6477-Datensätzen.
Abb. 4. Identifizierung von DEGs in den GSE6477-Datensätzen.
Fazit
Das multiple Myelom (MM) in China weist eine niedrige Überlebensrate auf. Die Sequenzierung von 50 Patienten ergab Mutationen in 337 Genen, insbesondere in MUC16, MUC4 und TTN. Wichtige Mutationen in BCL6 und BIRC3 sind mit schlechteren Ergebnissen und immunologischen Veränderungen verbunden, was neue Behandlungsziele bietet, jedoch weitere Forschung erfordert.
Referenz
- Xie C, Zhong L, Luo J, et al. Identifizierung von Mutationsgen-Prognose-Biomarkern bei multiplem Myelom durch Genpanel-Exomsequenzierung und Transkriptomanalyse in der chinesischen Bevölkerung. Computer in Biologie und Medizin, 2023, 163: 107224.
Verwandte Publikationen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Unterschiedliche Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die durch Therapie induzierte Seneszenz.
Journal: Zellsterben & Krankheit
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Zeitschrift: Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Erzeugung eines hochattenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genom-Analyse und Replikationsstudien des Simian Foamy Virus Serotyp 3 Stamm FV2014 des Afrikanischen Grünen Affen
Journal: Viren
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


