
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Gezielte Bisulfid-Sequenzierung
Als führender Anbieter von NGS Als Partner von Illumina ist CD Genomics darauf spezialisiert, ein Portfolio von Lösungen für epigenetische Studien anzubieten. Gekennzeichnet durch geringe Probenanforderungen, hohe Auflösung, Effizienz und Wirtschaftlichkeit, stellt das gezielte Bisulfid-Sequencing ein praktikables Werkzeug zur Analyse des Methylierungsstatus gezielter genomischer Regionen dar. Mit über 10 Jahren Berufserfahrung können wir Ihre Projektanforderungen und Budgets bei der Erforschung des Methyloms vollständig erfüllen.
Die Einführung der gezielten Bisulfit-Sequenzierung
DNA-Methylierung, die am häufigsten an Cytosinen in den CpG-Stellen vorkommt, spielt eine wichtige Rolle bei der Genexpression und -regulation. Die Fähigkeit, DNA-Methylierung präzise und effizient zu erkennen und zu bewerten, trägt dazu bei, unser Verständnis der DNA-Methylierung in Entwicklung und Krankheit zu verbessern. Während die Ganzgenom-Bisulfid-Sequenzierung methyliertes Cytosin auf einer Ganzgenom-Ebene identifiziert, ist die gezielte Bisulfid-Sequenzierung eine genaue, effiziente und wirtschaftliche Technologie zur Analyse der DNA-Methylierung in Zielregionen, die einen hybridiierungsbasierten Schritt auf Plattformen mit vorab entworfenen Oligos umfassen kann, die die CpG-Inseln, Genpromotoren und andere bedeutende methylierten Regionen erfassen, oder einen PCR-basierten Schritt, um mehrere bisulfid-konvertierte DNA-Regionen in einer einzigen Reaktion zu amplifizieren. Es sind kommerziell entworfene Capture-Bibliotheken mit einer Reihe von epigenetischen Merkmalen erhältlich, die etwa 12% bis 24% aller CpGs im Genom abdecken. Darüber hinaus werden spezifische Primer entworfen, um die Region von Interesse zu erfassen und standortspezifische DNA-Methylierungsänderungen zu bewerten.
Vorteile der gezielten Bisulfit-Sequenzierung
- Einzelbasenauflösende DNA-Methylierungsmusterung selektiver Regionen.
- Erhöhte Genauigkeit und Empfindlichkeit bei gleichzeitiger Senkung Ihrer Gesamtkosten.
- Ermöglicht eine bessere Bestimmung sowohl von SNPs als auch von Methylierungsstatusereignissen.
- Ein besseres Verständnis von Entwicklung und Krankheit bieten.
Anwendung der gezielten Bisulfit-Sequenzierung
- Forschung zu Mechanismen der Genregulation
- Methylierungsmarker-Screening
- Methylierungsvalidierung
- Tier- und Pflanzenzüchtungsforschung
- Klinische großangelegte Multi-Standort-Methylierungsstudien
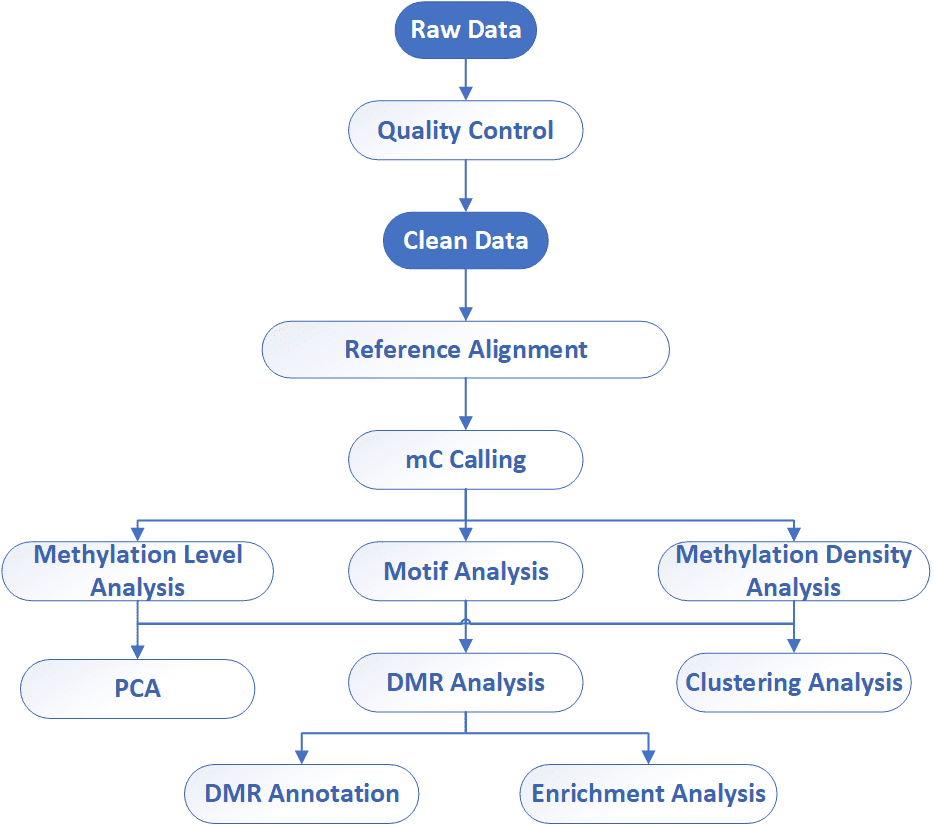
Gezielte Bisulfid-Sequenzierung-Workflow
Unser hochqualifiziertes Expertenteam und die strenge Qualitätskontrolle nach jedem Verfahren gewährleisten umfassende und genaue Ergebnisse. Im Prozess der gezielten Bisulfit-Sequenzierung wird die genomische DNA bisulfitkonvertiert und mit spezifisch entworfenen und validierten Primern oder Sonden multiplex amplifiziert. Die Amplicons werden durch Barcoding und Adapterisierung zusammengeführt. Die Amplicon-Bibliotheken werden dann mit Illumina HiSeq-Plattformen sequenziert.

Dienstspezifikationen
Musteranforderungen und Vorbereitung
|
|
 |
Sequenzierung
|
 |
Datenanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur gezielten Bisulfid-Sequenzierung für Ihre Schreibweise (Anpassung)
CD Genomics bietet hochwertige Daten und integrierte bioinformatische Analyse-Dienstleistungen für gezielte Bisulfit-Sequenzierung an, einschließlich Bisulfit-Konversion, Primer-/Probe-Design und Validierung, Hybridisierung/PCR-Amplifikation, Bibliotheksvorbereitung, DNA-Sequenzierung und Datenanalyse. CD Genomics verwendet sowohl kommerziell entworfene Capture-Arrays als auch maßgeschneiderte Capture-Bibliotheken, um Ihre Forschungsziele vollständig zu erfüllen. Bitte zögern Sie nicht, uns für weitere Informationen zu kontaktieren. Wir freuen uns auf eine Zusammenarbeit mit Ihnen in naher Zukunft.
Referenz:
- Ziller M. J. et al. Zielgerichtete Bisulfit-Sequenzierung des dynamischen DNA-Methyloms. Epigenetik & Chromatin, 2016, 9(1): 55.
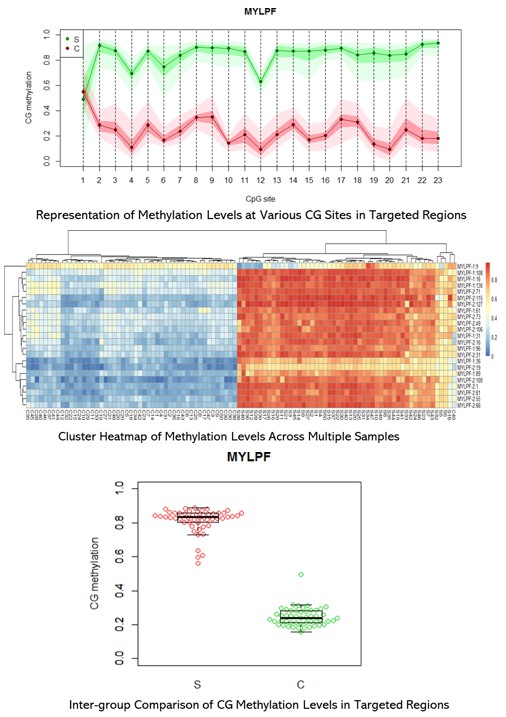
Demonstrationsergebnisse
Häufig gestellte Fragen zu gezielter Bisulfit-Sequenzierung
1. Wann sollten Sie gezielte Bisulfite-Sequenzierung wählen?
Die Technologie der gezielten Bisulfit-Sequenzierung kombiniert die Bisulfit-Umwandlung mit der gezielten Amplifikation von Regionen von Interesse, und die Next-Generation-SequenzierungEs ist ein schnelles und effizientes Werkzeug zur Analyse von bis zu 10 kb gezielter genomischer Regionen in bis zu 96 Proben gleichzeitig. Die Ergebnisse bieten eine absolute Quantifizierung der Cytosin-Methylierung mit einer Einzelbasenauflösung. Darüber hinaus kann diese Methode verwendet werden, um die Ergebnisse zu bestätigen, die aus Whole-Genome-Bisulfid-Sequenzierung, reduzierte Repräsentation Bisulfid-Sequenzierungund methylierte DNA-Immunpräzipitations-SequenzierungGezielte Bisulfit-Sequenzierung ermöglicht eine bessere Bestimmung von SNPs und Methylierungsstatusereignissen sowie ein besseres Verständnis menschlicher Krankheiten.
2. Was sind die Vorteile der gezielten Bisulfite-Sequenzierung im Vergleich zu anderen Technologien der Methylierungssequenzierung?
Im Vergleich zu Studien zur Methylierungsprofilierung des gesamten Genoms ist die Methylierungsdetektion nur ausgewählter Kandidatenregionen ein praktikablerer Ansatz, der die Methylierungsanalyse in verschiedenen Umgebungen machbarer und zugänglicher macht. Die gezielte Bisulfit-Sequenzierung erhöht die Datenrate erheblich und senkt die Kosten. Gleichzeitig ist sie auch in der Lage, den Methylierungsstatus von Regionen zu erfassen, die nicht detektiert werden können von RRBS (reduzierte Repräsentations-Bisulfid-Sequenzierung) oder Immunopräzipitation Ansätze. Zielgerichtete Bisulfit-Sequenzierung wurde umfassend auf die Methylierungssequenzierung und Validierung großer Stichprobenkohorten in Zielregionen angewendet.
3. Wie entwirft man geeignete Sonden oder Primer?
Eine erfolgreiche Anwendung der gezielten Bisulfit-Sequenzierung hängt weitgehend vom richtigen Design der Sonden und Primer ab. Es gibt viele effektive Werkzeuge zur automatischen Erstellung von Sonden oder Primern, wie ppDesigner (http://genome-tech.ucsd.edu/public/Gen2_BSPP/ppDesigner/ppDesigner.php) und PRIMEGENSw3 (http://primegens.org). Nach dem Erhalt vieler Sonden- oder Primer-Kandidaten ist der Validierungsprozess wichtig, um die optimalsten auszuwählen.
4. Wie ist der Arbeitsablauf der gezielten Bisulfit-Sequenzierung?
Der Arbeitsablauf der PCR-basierten gezielten Bisulfit-Sequenzierung ist in Abbildung 1 dargestellt. Bei der hybridisierungsbasierten gezielten Bisulfit-Sequenzierung kann entweder eine Bisulfit-Konversion von hybrid-selektiertem nativen DNA oder eine Hybridselektion von konvertierter DNA durchgeführt werden. Letzterer Ansatz ist mittlerweile kommerziell verfügbar und zeichnet sich durch eine überlegene Zielgenauigkeit, geringere DNA-Eingabebedürfnisse und die Fähigkeit aus, eine C-zu-U-Bisulfit-Konversion von einem C-zu-T-SNP zu unterscheiden.
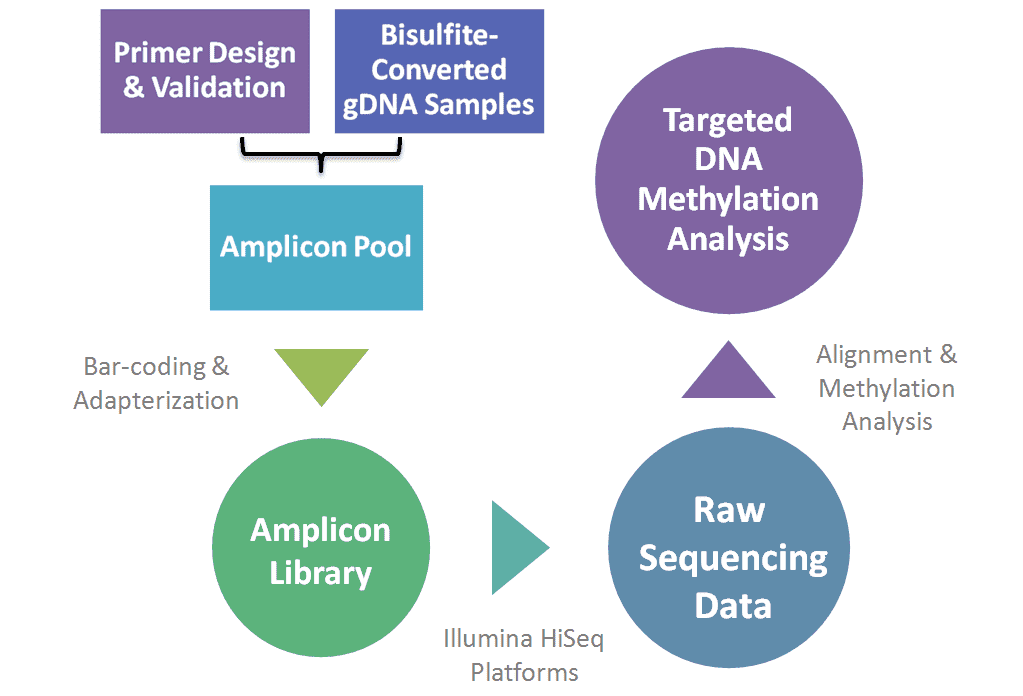 Abbildung 1. Der Arbeitsablauf der PCR-basierten gezielten Bisulfit-Sequenzierung.
Abbildung 1. Der Arbeitsablauf der PCR-basierten gezielten Bisulfit-Sequenzierung.
Gezielte Bisulfid-Sequenzierungs-Fallstudien
Gezielte Methylierungssequenzierung zeigt dysregulierte Wnt-Signalgebung bei Parkinsonkrankheit
Zeitschrift: Zeitschrift für Genetik und Genomik
Impactfaktor: 4,051
Veröffentlicht: 20. Oktober 2016
Hintergrund
Umwelt- und genetische Faktoren spielen eine entscheidende Rolle in der Ätiologie der Parkinson-Krankheit. Bis jetzt haben wir verstanden, dass eine Reihe von Umweltschadstoffen das Parkinson-Syndrom bei Menschen und Tieren begünstigen. Dennoch haben wir keine Ahnung von der potenziellen Wechselwirkung zwischen Umwelt und Genetik in der Pathogenese der Parkinson-Krankheit. Daher wenden Zhang et al. sowohl gezielte Bisulfit-Sequenzierung als auch immunhistochemische Methoden an, um die zugrunde liegenden Zusammenhänge zu untersuchen.
Methoden
- Gefrorene Gewebe
- Formalin-fixierte Gewebe
- Genomische DNA-Extraktion
- Immunfärbung
- Gezielte DNA-Erfassung
- Shotgun-Bibliothekskonstruktion
- Bisulfit-Padlock-Sequenzierung
- Expressionsprofilierung Array-Analyse
- Ganzgenom-Expressionsprofilierung
Ergebnisse
Gezielte Bisulfit-Sequenzierung zeigte eine erhöhte Methylierung von Genen im Wnt-Signalweg.
Sie entwarfen ein Set von etwa 97.000 Vorhängeschloss-Proben, die 16.379 validierte Gewebe-differenzielle Methylierungsregionen (T-DMRs) abdeckten, alle bekannten und vorhergesagten menschlichen imprinting Gene sowie die Transkriptionsstartstellen aller Gene auf dem X-Chromosom. Die K-Means-Clustering-Analyse ergab 336 nicht überlappende DMRs, von denen einige bekannten Genen zugeordnet wurden. Die Mehrheit dieser Regionen (230/233) zeigte im Vergleich zu gesunden Spendern erhöhte Methylierungsniveaus in den Gehirngeweben von PD. Es gibt eine signifikante Anreicherung von Genen, die an der Wnt-Signalübertragung und der Neurogenese beteiligt sind.
2. Verminderte Detektion von Genen im Wnt-Signalweg in der Substantia nigra von PD-Patienten
Die Werte von Foxc1, Ngn2, Spry1 und β-Catenin in DA-Neuronen des Mittelhirns waren bei PD-Patienten im Vergleich zu den entsprechenden Kontrollen deutlich reduziert. Die quantitative Analyse ergab einen signifikanten Anstieg der Zellen mit reduzierter oder nicht nachweisbarer Expression dieser Gene. Im Gegensatz dazu blieb der NEDD8-Spiegel in den DA-Neuronen sowohl bei PD-Patienten als auch bei Kontrollen ähnlich, was darauf hindeutet, dass die reduzierte Nachweisbarkeit dieser Proteine in den DA-Neuronen des Mittelhirns bei PD-Patienten spezifisch ist. Ähnliche Beobachtungen wurden zu Foxc1 und Spry1 in den kortikalen Geweben von PD-Patienten und ihren entsprechenden Kontrollen gemacht. Allerdings wurden in den kortikalen Geweben von PD-Patienten und ihren entsprechenden Kontrollen nur geringe Veränderungen von Ngn2 und β-Catenin festgestellt.
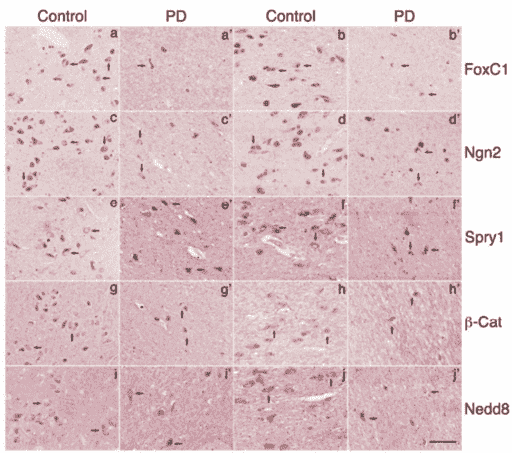 Abbildung 1. Die Immunodetektion von Foxc1, Ngn2, Spry1, β-Catenin und Nedd8 im menschlichen Mittelhirn.
Abbildung 1. Die Immunodetektion von Foxc1, Ngn2, Spry1, β-Catenin und Nedd8 im menschlichen Mittelhirn.
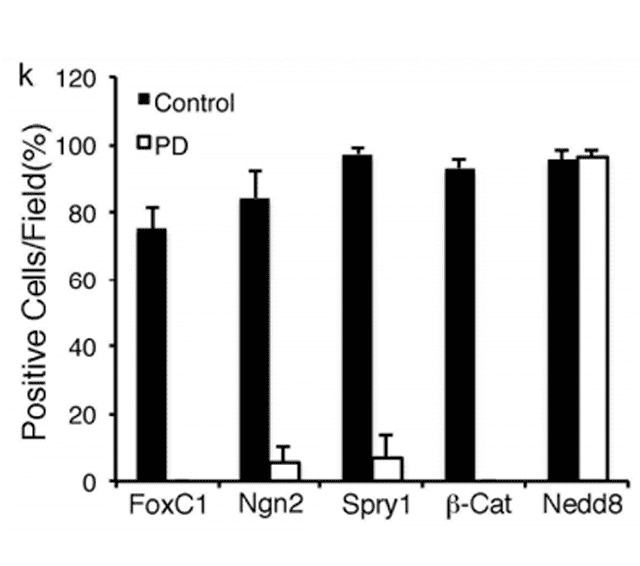 Abbildung 2. Die quantitative Analyse zeigte einen signifikanten Anstieg von Zellen mit reduzierter oder nicht nachweisbarer Expression dieser Gene.
Abbildung 2. Die quantitative Analyse zeigte einen signifikanten Anstieg von Zellen mit reduzierter oder nicht nachweisbarer Expression dieser Gene.
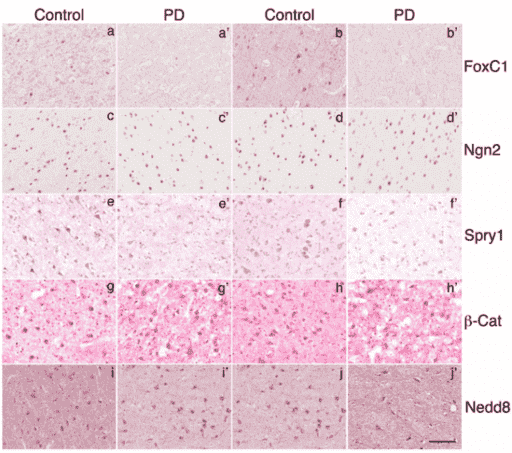 Abbildung 3. Die Immunodetektion von Foxcl, Ngn2, Spry1, β-Catenin und Nedd8 im menschlichen Gehirnrinde.
Abbildung 3. Die Immunodetektion von Foxcl, Ngn2, Spry1, β-Catenin und Nedd8 im menschlichen Gehirnrinde.
3. Herunterregulierung der Wnt-Signalgebung in Zellen, die mit dem PD-Neurotoxin MPP+ behandelt wurden
Die funktionelle Annotationsanalyse durch MPP+-Behandlung zeigt eine Anreicherung von hochregulierten Genen, die in Disulfidbindungen, Zellmembran, neurologischen Systemprozessen, Abwehrreaktionen, Zelladhäsion, positiver Regulation der Apoptose und Regulation der Migration tätig sind. Zu den durch MPP+ induzierten herunterregulierten Annotationsclustern gehören Gene, die in Disulfidbindungen, Sekretion, extrazellulärer Matrix, Plasmamembran, Wnt-Signalgebung und Hedgehog-Signalgebung tätig sind. Die Ergebnisse unterstützen weiter die Vorstellung, dass die Wnt-Signalgebung bei neurotoxininduzierter Parkinsonismus beeinträchtigt ist.
Referenz:
- Zhang L, Jie D, Qian P, et al. Zielgerichtete Methylierungssequenzierung zeigt dysregulierte Wnt-Signalgebung bei Parkinson-Krankheit. Zeitschrift für Genetik und Genomik, 2016, 43(10):587-592.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Naturmikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft verursachen Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Internationales Journal für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die duale Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nucleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


