
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Genetische Verknüpfungskarte
Um den aufkommenden Bedürfnissen der Forschungsgemeinschaften gerecht zu werden, hat CD Genomics einen erschwinglichen, zuverlässigen Service für genetische Verknüpfungskarten entwickelt, der auf Hochdurchsatz-Sequenzierung dichte Marker zu erhalten und den Forschern eine hochwertige genetische Verknüpfungskarte sowie professionelle Datenanalysen zu bieten.
Die Einführung der genetischen Verknüpfungskarte
Genetische Verknüpfungskarte, auch bekannt als genetische Karte, ist ein lineares Diagramm der Sequenz und relativen Abstände von molekularen Markern auf Chromosomen, basierend auf den Häufigkeiten von Rekombinationen zwischen Markern während des Crossing-overs homologer Chromosomen. Der Aufbau einer hochdichten genetischen Karte basiert auf Hochdurchsatz-Sequenzierung Die Technologie hat sich allmählich zu einer revolutionären Technologie entwickelt, die von Forschern bevorzugt wird. Sie kann in kurzer Zeit eine große Anzahl von molekularen Markern entwickeln und eine ultra-hochdichte genetische Karte erstellen. Sie liefert genaue und vollständige Informationen über die Anzahl der QTLs und deren Loci, die mit dem Phänotyp ko-segregieren.
Wenn Sie mehr über genetische Verknüpfungskarten erfahren möchten, können Sie unseren Artikel lesen "Genetische Verknüpfungsanalyse: Definition, Techniken und Anwendungen".
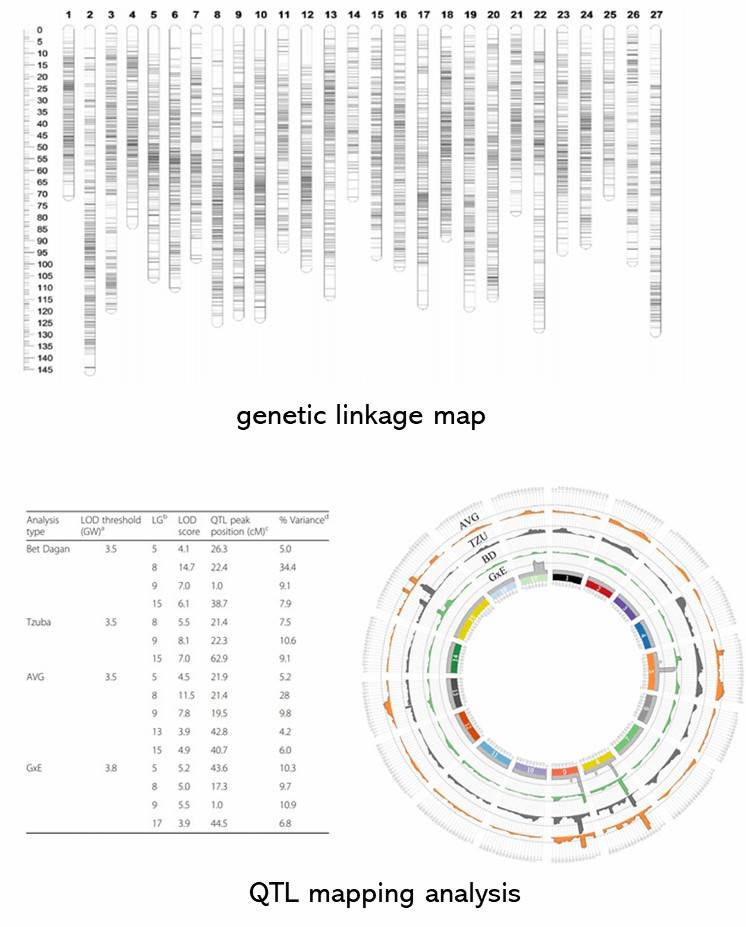
 Abbildung 1. Eine genetische Verknüpfungskarte (Yuhui Zhao et al. 2020)
Abbildung 1. Eine genetische Verknüpfungskarte (Yuhui Zhao et al. 2020)
Was sind die Vorteile einer genetischen Verknüpfungskarte?
- Verständnis der genetischen Vererbung
- Genidentifikation und -kartierung
- Marker-gestützte Selektion
- Studien zur genetischen Vielfalt
- Erleichterung der QTL-Analyse
- Genomforschung und -entwicklung
- Verbesserung der Pflanzen- und Tierproduktion
- Unterstützung für funktionelle Genomik
- Verbesserung der genetischen Beratung und Krankheitsvorhersage
- Fortschritte in der personalisierten Medizin
- Hohe Geschwindigkeit, hohe Dichte, hohe Qualität
- Hochwertige Sequenzierungsdaten, Hochleistungsrechnerplattform
Was sind die Anwendungen von genetischen Verknüpfungskarten?
 Abbildung 1. Eine genetische Verknüpfungskarte (Yuhui Zhao et al. 2020)
Abbildung 1. Eine genetische Verknüpfungskarte (Yuhui Zhao et al. 2020)
Genetische Verknüpfungskarten-Workflow

Dienstspezifikation
Musteranforderungen
|
|
 |
Sequenzierung
|
 |
Bioinformatikanalyse
Wir bieten maßgeschneiderte bioinformatische Analysen an, einschließlich:
|
Analyse-Pipeline
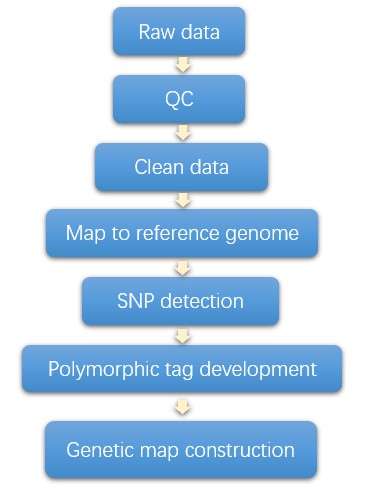
Liefergegenstände
- Rohdaten (FASTQ)
- Markerinformationen
- Bewertungsbericht zur genetischen Verknüpfungskarte
- Datenanalysebericht
Referenz:
- Zhao YH, u. a.Hochdichte genetische Verknüpfungskartenkonstruktion und QTL-Kartierung für wichtige Fruchteigenschaften. PLoS ONE2020,15(2): e0229020.
Demo-Ergebnisse
Häufig gestellte Fragen zum genetischen Verknüpfungsdiagramm
1. Was ist eine genetische Verknüpfungskarte?
Im Wesentlichen ist eine genetische Verknüpfungskarte eine diagrammatische Darstellung, die die relativen Positionen von Genen oder genetischen Markern auf einem Chromosom darstellt, abgeleitet aus ihren jeweiligen Rekombinationsfrequenzen. Diese Art von Karte bietet einen unverzichtbaren visuellen Leitfaden für die Reihenfolge und den Abstand zwischen genetischen Loci. Sie dient dem doppelten Zweck, die Komplexität der genetischen Vererbung zu beleuchten und praktikable Ansätze in so unterschiedlichen Bereichen wie Pflanzenzüchtung, Genkartierung und umfassender genetischer Forschung zu informieren.
2. Wie wird eine genetische Verknüpfungskarte erstellt?
Die Erstellung einer genetischen Verknüpfungskarte folgt im Allgemeinen der Reihenfolge der unten aufgeführten Schritte:
- Auswahl einer geeigneten Population: Häufig gewählte Populationen bestehen aus F2-, Rückkreuzungs-Populationen oder rekombinanten inbred Linien.
- Genotypisierung Implementierung: Die Identifizierung genetischer Marker nutzt Technologien wie SNP-Arrays oder umfassende Whole-Genome-Resequenzierung.
- Datenverarbeitung und analytische Auswertung: Durch die Nutzung sowohl computergestützter Werkzeuge als auch statistischer Methoden werden Rekombinationsfrequenzen zwischen Markern berechnet, was die Erstellung von Kopplungsgruppen und anschließend den Aufbau des genetischen Karten ermöglicht.
3. Wie sollten Elterntiere ausgewählt werden?
Die Auswahl der Elterntypen beeinflusst direkt den Schwierigkeitsgrad beim Kartenbau und den Anwendungsbereich der erstellten Karte. Sie muss die folgenden Kriterien erfüllen: (i) Genetische Polymorphie zwischen den Elterntypen. (ii) Berücksichtigung der Fruchtbarkeit der Nachkommen, um eine verzerrte Segregation zu verhindern. (iii) Hochreine Elterntypmaterialien sollten nach Möglichkeit ausgewählt werden (außer der F1-Generation).
4. Welche Software-Tools gibt es zum Erstellen von Karten?
JoinMap ist ein Softwaretool, das auf dem Windows-Betriebssystem läuft und derzeit am weitesten verbreitet ist, da es für nahezu alle Populationstypen anwendbar ist. Weitere Tools sind R/qtl, Lepmap, Highmap, Onemap, Mstmap und Carthagen.
5. Was ist die Rekombinationsfrequenz und warum ist sie wichtig?
Tatsächlich dient die Rekombinationsfrequenz als ein wesentliches Parameter, wenn es darum geht, die zugrunde liegende Genetik eines Organismus zu umreißen. Sie bezieht sich auf den Prozentsatz der Nachkommen, bei denen ein spezifisches Crossing-over-Ereignis zwischen zwei Genen oder Markern während des Prozesses der Meiose stattgefunden hat. Offensichtlich deutet eine höhere Rekombinationsfrequenz auf eine größere genetische Distanz hin, die die betreffenden Gene trennt. Die Bedeutung dieser Frequenz liegt in ihrer unverzichtbaren Rolle beim Aufbau genetischer Verknüpfungskarten - diese Frequenzen helfen dabei, die relativen Positionen der Gene innerhalb eines Chromosoms zu bestimmen. Daher trägt die Rekombinationsfrequenz erheblich zu unserem Verständnis und zur Identifizierung der Genarchitektur und -funktion bei.
6. Was sind die Maßstäbe für die Kartenqualität?
Die Qualitätsbestimmung von Karten basiert hauptsächlich auf Indikatoren wie statistischer Kartierung, Kollinearitätsanalyse von genetischen und physischen Karten sowie der Analyse von Rekombinations-Hitzekarten benachbarter Marker.
7. Wie unterscheidet sich eine genetische Verknüpfungskarte von einer physikalischen Karte?
Eine genetische Verknüpfungskarte ist nach den Rekombinationsfrequenzen organisiert und zeigt die relativen Positionen von Genen oder Markern auf einem Chromosom. Im Gegensatz dazu wird eine physikalische Karte basierend auf den genauen physischen Standorten und definitiven Abständen von Genen auf Chromosomen erstellt, die durch die tatsächliche DNA-Sequenz bestimmt werden. Die Integration dieser beiden Kartentypen bietet ein umfassendes Verständnis der genomischen Struktur und Funktion.
Genetische Verknüpfungskarten Fallstudien
Eine ultra-hochdichte genetische Karte liefert Einblicke in die Genom-Syntenie, die Rekombinationslandschaft und die Hautfarbe der Hauptwurzel bei Rettich.Radieschen L.)
Journal: Pflanzenbiotechnologie-Journal
Impactfaktor: 13,263
Veröffentlicht: 20. Juni 2019
Hintergrund
Hochdichte genetische Karten sind entscheidend für genetische und genomische Forschungen, einschließlich der Kartierung quantitativer Merkmalslokalisationen (QTLs) in Pflanzen. Die Auflösung der QTL-Kartierung hängt von der Marker-Dichte und der Populationsgröße ab, und Hochdurchsatz-Nachsequenzierung verbessert die Markerentwicklung und GenotypisierungAnthocyane, die für die Farben in verschiedenen Pflanzen verantwortlich sind, bieten gesundheitliche Vorteile und spielen eine Rolle bei der Anziehung von Bestäubern und dem Schutz. Die meiotische Rekombination, die entscheidend für die Schaffung neuer Allelkombinationen ist, beeinflusst die genomische Evolution und die Pflanzenzüchtung. Hochdichte genetische Karten helfen, die Verteilung der Rekombination und Hotspots zu untersuchen. Bei Radieschen wurde eine hochdichte Kopplungskarte erstellt, die QTLs und Kandidatengene für wichtige Merkmale identifiziert und Einblicke für genetische Verbesserungen und Studien zur Genom-Evolution bietet.
Methoden
- Pflanzenmaterialien
- Phänotypdatenaufnahme
- Zwei fortgeschrittene Radieschen-Inzuchtlinien
- Populationsresequenzierung
- Genotypisierung
- Illumina HiSeq 2500 Plattform
- Phänotypbewertung
- Bin-Kartenkonstruktion
- QTL-Kartierung
- RT-qPCR-Analyse
Ergebnisse
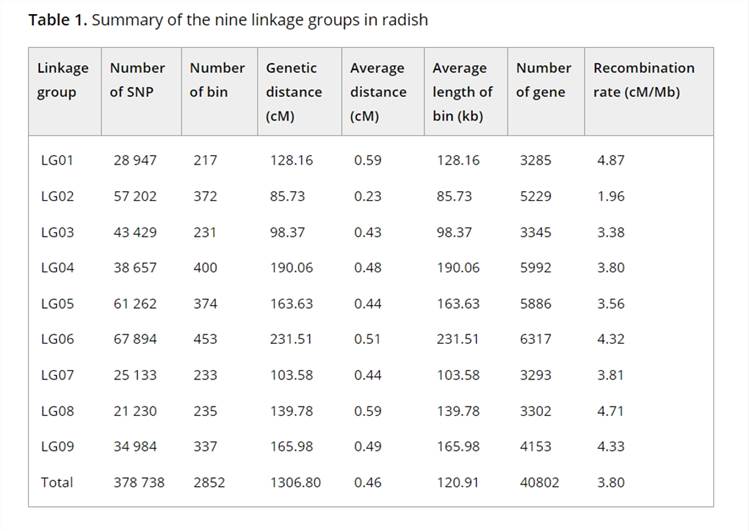
Um eine hochauflösende genetische Verknüpfungskarte im Rettich zu erstellen, wurde eine Ganzgenom-Nachsequenzierung von 137 F2-Individuen und ihren Elternlinien unter Verwendung der Illumina HiSeqTM 2500-Plattform durchgeführt. Dies ergab 403 Gb an sauberen Reads, die auf das Referenzgenom des Rettichs abgebildet wurden, und identifizierte nach der Filterung 411.891 SNPs. Mit einem gleitenden Fensteransatz wurden 2.852 Rekombinations-Bin-Marker erstellt, die eine genetische Distanz von 1.306,8 cM abdeckten, mit einer durchschnittlichen Distanz von 0,46 cM zwischen den Markern. In einigen Markern wurde eine signifikante Segregationsverzerrung beobachtet, wodurch 19 Segregationsverzerrungsregionen über sieben Verknüpfungsgruppen identifiziert wurden. Verzerrte Marker wurden beibehalten, um die Abdeckung der Verknüpfungsgruppen zu verbessern.
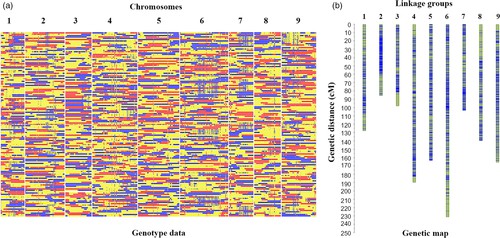 Abb. 1. Rekombinations-Bin-Karte (a) und genetische Karte (b) von 137 F2-Individuen.
Abb. 1. Rekombinations-Bin-Karte (a) und genetische Karte (b) von 137 F2-Individuen.
In der F2-Population wurde die Erkennung von 17 quantitativen Merkmal-Loci (QTLs) festgestellt, von denen sieben mit zuvor anerkannten QTLs in derselben Kopplungsgruppe übereinstimmten. Die markierten QTL-Regionen hatten im Durchschnitt eine Länge von 168 Kilobasen und erstreckten sich über fünf verschiedene Kopplungsgruppen. Die Methode, die zur Isolierung des Gens führte, das die Wurzelhautfarbe beeinflusst, umfasste einen Kreuzung zwischen den Sorten 'NAU-LB' und 'NAU-YH'. Die Ergebnisse deuteten darauf hin, dass die Dominanz des sogenannten R-Gens für die Regulierung der roten Hautfarbe verantwortlich ist. Die spezifische Lokalisation des R-Gens wurde auf eine Region von 72 Kilobasen auf Chromosom 7 eingegrenzt. Weitere Untersuchungen identifizierten das RsMYB90-Gen als das wahrscheinliche Gen, das an der Kontrolle der roten Hautfarbe beteiligt ist. Es zeigte erhöhte Expressionsniveaus, die hauptsächlich in den rothäutigen Sorten vorkamen. Interessanterweise weist das RsMYB90-Gen einen bemerkenswerten Grad an Homologie mit Mitgliedern der MYB-Transkriptionsfaktor-Familie auf, einer Gruppe, die für ihre Beteiligung am Prozess der Anthocyanin-Biosynthese bekannt ist.
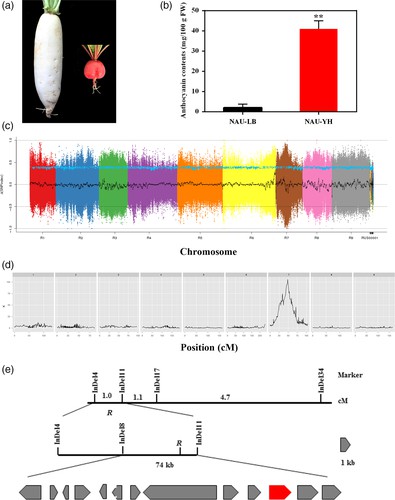 Abb. 2. Kartenbasierte Klonierung des roten Hautgens R.
Abb. 2. Kartenbasierte Klonierung des roten Hautgens R.
Die Positionen der Bin-Marker auf der genetischen Karte wurden mit ihren physischen Positionen im Radieschen-Genom-Assembly verglichen. Alle Bin-Marker wurden auf die neun Pseudo-Chromosomen kartiert, die 80,7 % des 424 Mb umfassenden Referenzgenoms des Radieschens abdecken. Während eine hohe Kollinearität zwischen der genetischen Karte und den Chromosomen festgestellt wurde, wurden einige Inkonsistenzen bemerkt. Insbesondere waren die Bins bei 26–43 Mb auf Chromosom 2 invertiert, und die Reihenfolge der Bins an den distalen Enden der Chromosomen 1, 3 und 4 stimmte nicht mit der genetischen Karte überein.
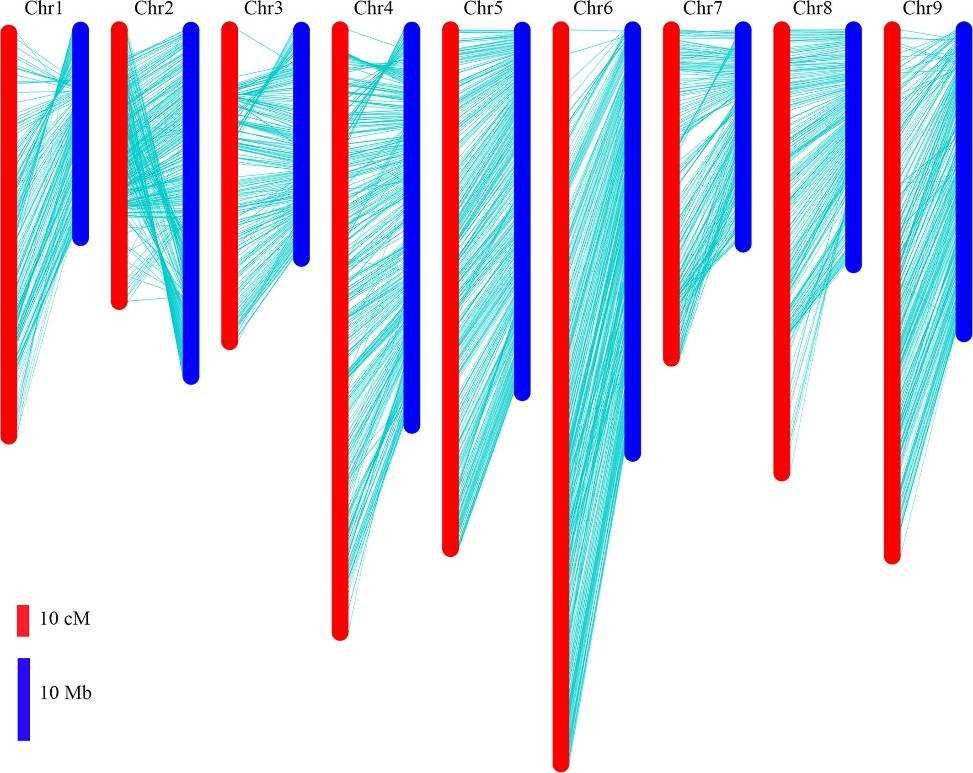 Abb. 3. Vergleich zwischen der genetischen Karte und der Radieschen-Genomsequenz.
Abb. 3. Vergleich zwischen der genetischen Karte und der Radieschen-Genomsequenz.
Fazit
Durch Whole-Genome-SequenzierungWir haben eine hochauflösende, hochpräzise dichte genetische Karte erstellt. In Kombination mit der Quantitative Trait Locus (QTL)-Kartierung und der positionalen Klonierung wurde RsMYB90 als Kandidatengen identifiziert, das die rote Haut von Radieschen-Rüben steuert. Das RsMYB90-Gen spielt eine entscheidende Rolle bei der Regulierung der Anthocyanin-Biosynthese und liefert Einblicke in die genetische Regulation der Anthocyanin-Akkumulation in Radieschen.
Referenz:
- Luo X, Xu L, Wang Y, u. a.Eine ultra-hochdichte genetische Karte liefert Einblicke in die Genom-Syntenie, die Rekombinationslandschaft und die Hautfarbe der Hauptwurzel bei Rettich.Radieschen L.). Pflanzenbiotechnologie-Journal, 2020, 18(1): 274-286.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien bei Aymara, Quechua und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP) Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Journal: JAMA Netzwerk Offen
Jahr: 2023
Erhöhte Produktion von pathogenen, luftgetragenen Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen
Journal: Mikrobiologie Spektrum
Jahr: 2023
Eine Spleißvariante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Zeitschrift: Zellen
Jahr: 2021
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


