
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
![]()
Was ist metatranskriptomische Sequenzierung?
Metatranskriptomische Sequenzierung bietet einen Echtzeit-Einblick in die Genaktivität innerhalb mikrobieller Gemeinschaften. Anstatt zu analysieren, welche Gene vorhanden sind (wie in der Metagenomik), konzentriert sich diese Methode darauf, welche Gene unter bestimmten Bedingungen aktiv exprimiert werden – und offenbart mikrobielles Verhalten, Regulation und metabolische Ausgaben. Egal, ob Sie das Mikrobiom des Darms oder ein industrielles Fermentationssystem untersuchen, die Metatranskriptomik beantwortet die Frage:
„Was machen die Mikroben gerade?“
Wie es funktioniert:
- Extrahiere totale RNA aus der Probe (z. B. Stuhl, Boden, Gewebe)
- Entfernen Sie ribosomale RNA (rRNA), um die messenger RNA (mRNA) anzureichern.
- mRNA in komplementäres DNA (cDNA) umwandeln
- Konstruiere Sequenzierungsbibliotheken und führe Hochdurchsatz-Sequenzierung durch.
- Analysiere Genexpressionsmuster, um aktive mikrobielle Funktionen aufzudecken.
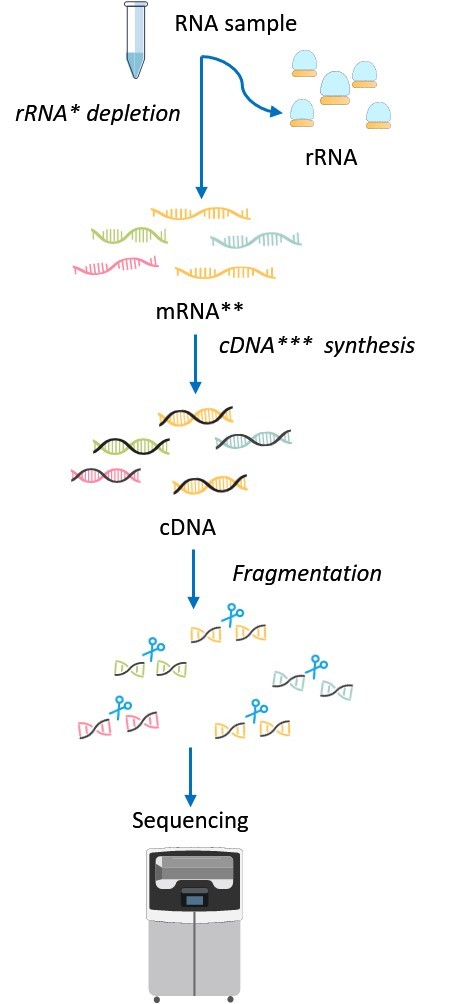 Metatranskriptomische Sequenzierungs-Workflow.
Metatranskriptomische Sequenzierungs-Workflow.
Warum Metatranskriptom-Sequenzierung wählen?
Diese Technik der nächsten Generation bietet tiefgehende funktionale Einblicke, die DNA-basierte Methoden nicht liefern können. Anstatt einfach nur zu identifizieren Wer ist da?, es offenbart was sie tun.
- Direkter Einblick in die mikrobielle Funktion
Quantifiziert die Genexpression über alle mikrobiellen Bereiche hinweg – Bakterien, Archaeen, Pilze und Viren. - Kulturfreie, umfassende Erkennung
Durch die Umgehung der Notwendigkeit von Kultivierung erfasst die Methode die Dynamik von Gemeinschaften in der realen Welt über alle Mikroben hinweg, einschließlich nicht kultivierbarer Arten. - Dynamische, zeitaufgelöste Analyse
Vergleichen Sie die Genexpression über Zeitpunkte oder Behandlungsbedingungen hinweg, um funktionale Veränderungen zu identifizieren oder potenzielle Biomarker zu erkennen. - Unterstützt mechanistische Entdeckung
Kombiniert mit Datenbanken wie KEGG hilft es, Stoffwechselwege und regulatorische Netzwerke zu rekonstruieren. - Kompatibel mit Multi-Omics
Integriert sich nahtlos mit Metagenomik, Wirts-Transkriptomik und Metabolomik für eine tiefere biologische Interpretation.

Wie schneidet es im Vergleich zu anderen Mikrobiom-Tools ab?
| Technik | Was es erkennt | Spiegelt funktionale Aktivität wider? | Auflösung | Am besten geeignet für |
|---|---|---|---|---|
| 16S/ITS Amplicon | Marker-Gene von Bakterien/Pilzen | ❌ Nein | Medium (Gattung/Art) | Schnelluntersuchung, taxonomische Profilierung |
| Metagenomik | Alle mikrobielle DNA (Taxonomie + potenzielle Funktionen) | ❌ Nein (nur funktionales Potenzial) | Hoch (Belastungsniveau) | Identifizierung von Arten und potenziellen Stoffwechselfähigkeiten |
| Metatranskriptomik | Aktiv exprimierte mikrobielle RNA | ✅ Ja | Hoch (Gen- + Stamm-Ebene) | Expressionsprofilierung, Mechanismusstudien, Biomarkerentdeckung |
| Gastgeber Transkriptomik | Wirt-RNA-Expression | ✅ Ja | Hoch | Studien zu Wirt-Mikrobe-Interaktionen |
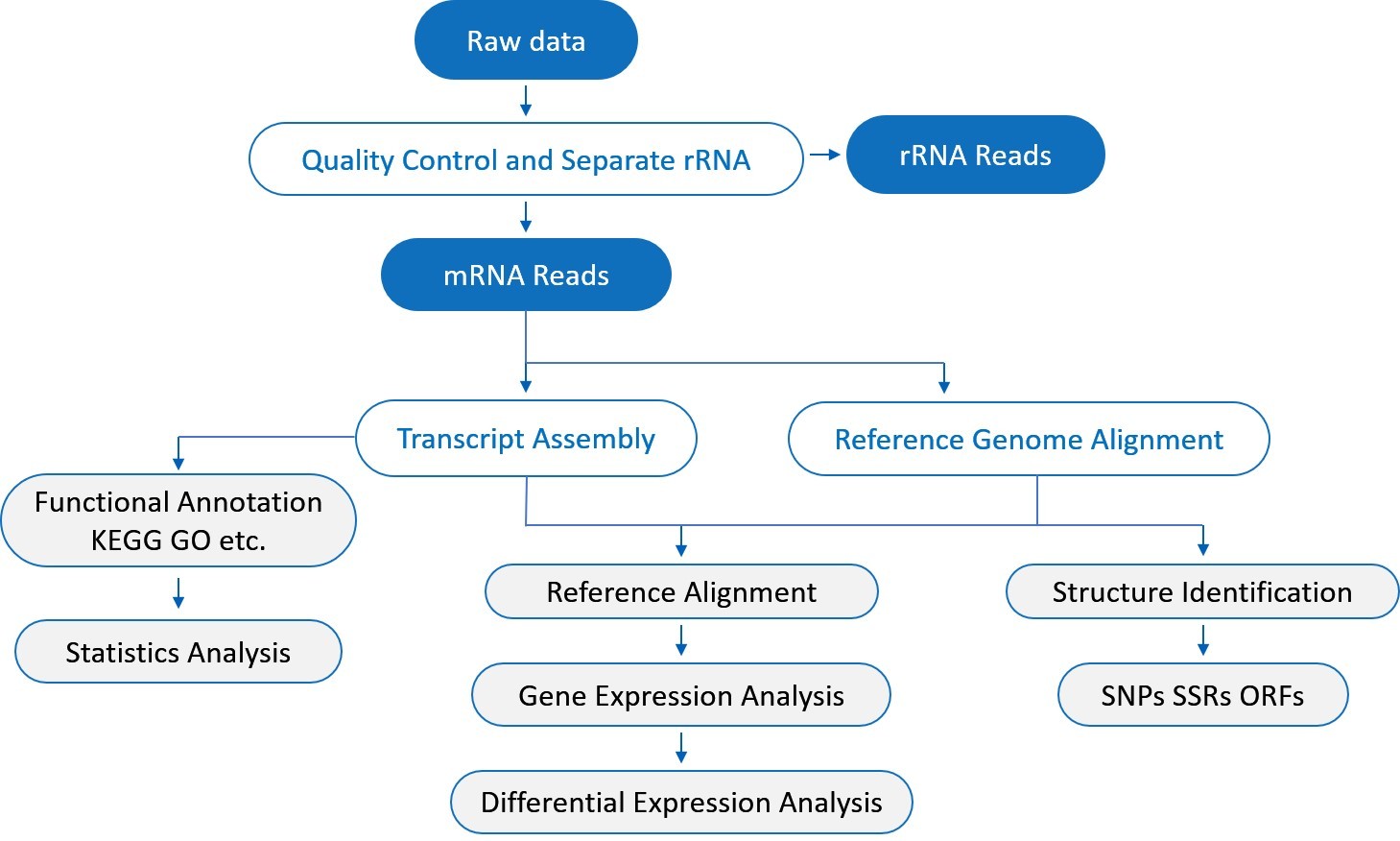
End-to-End Metatranskriptom-Sequenzierungsdienst-Workflow
Optimierter Service von der Probe bis zu den Ergebnissen – Maximierung von Qualität und Effizienz.

Ziele definieren
Workflow bestätigen

Proben registrieren
RNA-Qualitätskontrolle
(Optional) RNA-Extraktion

Entfernen Sie rRNA (>90%)
Erstellen Sie dual-indizierte Bibliotheken
Führen Sie die Qualitätskontrolle der Bibliothek durch.

Illumina / MGI Kurzlesungen
PacBio-Langsequenzen
Anpassbare Tiefe

Daten-QC
Transkriptomanalyse
Funktionale Annotation
Berichtserstellung und -lieferung
Überblick über die metatranskriptomische Sequenzierungsstrategie
Unterstützte Probenarten:
- Stuhl, Gewebe, Speichel, Abstriche und mehr
Höhepunkte des Bibliotheksbaus:
- >90% rRNA-Depletion
- Einzigartige doppelte Indizierung
- Strenge Qualitätsvalidierung
Sequenzierungsplattformen:
- Illumina NovaSeq X150 bp PE – breites Expressionsprofiling
- MGI DNBSEQ-G400100/150 bp PE – kosteneffektive Transkriptomik
- PacBio Sequel IIe15–25 kb HiFi – Isoform-Ebene Auflösung
Empfohlene Tiefe:
- Standard: 5–10 Gb/Stichprobe
- Optional: Höhere Tiefe für Transkripte mit niedriger Häufigkeit
Datenqualitätsmetriken:
- >80% Basen bei Q30+
- Fehlerquote <0,1%
- Genau, zuverlässige Ergebnisse
Metatranskriptomische Bioinformatikanalyse
Vollständig Datenanalyse Von Rohdaten zu publikationsreifen Visualisierungen, die Ihre Forschungs- und Förderbedürfnisse unterstützen.
Mikrobielle Expressionsprofilierung
- Profil der aktiven Genexpression über Bakterien, Pilze, Viren und Archaeen
- Visualisierung von Artenhäufigkeit und Diversitätsmetriken
Funktionale Annotation und Pfadanalyse
- Schlüsselgene mit UniRef und UniProt annotieren
- Rekonstruieren von Stoffwechselwegen mit KEGG und MetaCyc
- Führen Sie eine differenzielle Pfadexpressionsanalyse durch.
Antibiotikaresistenz- und Virulenznachweis
- Antibiotikaresistenzgene (AMR) und Virulenzfaktoren nachweisen
- Quantifizierung der Expression und Analyse der Pfadanreicherung
Veröffentlichungsbereite Visualisierungen und Berichte
- Hochwertige PCA, Heatmaps, Vulkanplots und mehr
- Statistische Analyse einschließlich LEfSe für wichtige funktionale Unterschiede
- Expressionsdaten normalisiert und in benutzerfreundlichen Tabellen bereitgestellt
Beispielanforderungen für metatranskriptomische Sequenzierung
Um eine optimale Sequenzierungsleistung zu gewährleisten, müssen Proben die grundlegenden Mengen- und Reinheitsstandards erfüllen. Eine individuelle Beratung ist für spezialisierte Probenarten verfügbar.
| Probenart | Mindestanforderungen |
|---|---|
| Gesamt-RNA | ≥ 4 μg (≥ 3 μg Minimum), ≥ 50 ng/μL |
| Kultivierte Zellen | ≥ 5 × 10⁶ Zellen |
| Umweltproben | ≥ 1,5 Gramm |
📩 Nicht sicher, ob Ihr Muster geeignet ist? Kontaktieren Sie uns für eine persönliche Beratung zur Vorbereitung.
Ist Metatranskriptomik das Richtige für meine Forschung?
Metatranskriptomische Sequenzierung zeigt, was Ihr Mikrobiom tatsächlich tut, nicht nur, wer dort ist. Wenn Ihre Studie mikrobielle Aktivität, Dynamik der Genexpression oder Veränderungen in funktionalen Wegen umfasst, könnte diese Technik perfekt geeignet sein.
Ideale Anwendungsfälle:
- Gesundheit & Krankheitsmikrobiomstudien
Verfolgen, wie sich die Mikrobiome des Darms, der Haut oder der Atemwege in gesunden oder kranken Zuständen verhalten. - Medikamenten-, Diät- oder Umweltinterventionen
Quantifizieren Sie, wie Behandlungen die mikrobielle Genexpression und Stoffwechselwege beeinflussen. - Umwelt- und Agrarische Mikrobielle Ökologie
Bewerten Sie die aktiven Funktionen von Mikroben in Böden, Gewässern oder wirtverbundenen Systemen. - Probiotika- und Mikrobiom-Therapeutika-Entwicklung
Identifizieren Sie nützliche Stämme und validieren Sie deren funktionale Mechanismen in Aktion. - Unkultivierbare Mikrobentdeckung
Aktive Expression von schwer kultivierbaren Organismen erkennen, die von traditionellen Methoden übersehen wurden.
Häufige Forschungsfragen, bei denen wir helfen können:
- Wie verändert sich die mikrobielle Genaktivität als Reaktion auf eine Behandlung oder Bedingung?
- Welche Gene oder Signalwege werden nach einer diätetischen oder pharmakologischen Intervention aktiviert?
- Kann ich neue Funktionen in Mikroben entdecken, die sich nicht im Labor kultivieren lassen?
- Wie kann ich Stämme funktional charakterisieren für die Entwicklung von Probiotika oder Biomarkern?
Kombinieren Sie Metatranskriptomik mit anderen Omics für tiefere Einblicke
| Gepaarte Technik | Kombinierter Vorteil | Beispielanwendung |
|---|---|---|
| Metagenomik + Metatranskriptomik | Identifizieren Sie sowohl potenzielle als auch tatsächliche Genaktivität. | Unterscheidung zwischen stillen und aktiven Stämmen in mikrobiellen Gemeinschaften |
| Gastgeber Transkriptomik Metatranskriptomik | Entschlüsselung von Wirt-Mikrobe-Interaktionsnetzwerken | Untersuchen von Entzündungs-/Infektionsmodellen |
| Metabolomik + Metatranskriptomik | Verknüpfen Sie die Genexpression mit dem tatsächlichen Stoffwechseloutput. | Untersuchen Sie den Einfluss von Medikamenten/Diäten auf den mikrobiellen Stoffwechsel. |
| 16S/ITS + Metatranskriptomik | Große Kohorten screenen und dann in aktive Proben hineinzoomen. | Effiziente Proben-Triage vor der tiefgehenden funktionalen Profilierung |
Warum CD Genomics für metatranskriptomische Sequenzierung wählen?
Wenn es darum geht, die mikrobielle Genexpression präzise und umfassend zu erfassen, bietet CD Genomics mehr als nur Sequenzierung – wir liefern umsetzbare Erkenntnisse, die auf jahrelanger Erfahrung und umfassendem Support basieren.
- Umfangreiche Multi-Omics-Expertise
Vertraut von führenden Forschungsinstituten und Biotech-Unternehmen bringen wir eine solide Erfolgsbilanz in den Bereichen Transkriptomik, Genomik und Mikrobiom-Profiling mit. - Flexible Plattformoptionen
Wählen Sie aus den Plattformen von Illumina, MGI oder PacBio, um Ihren Proben-Typ, Ihr Budget und Ihre Auflösungsbedürfnisse zu berücksichtigen. - Angepasste bioinformatische Analyse
Gewinnen Sie tiefere Einblicke durch fortgeschrittene Analysen, einschließlich funktioneller Annotation, Pfadanreicherung und differenzieller Expressionskartierung. - Strenge Qualitätskontrolle in jedem Schritt
Von der Probenbearbeitung bis zur endgültigen Berichterstattung gewährleistet unser End-to-End-Rückverfolgbarkeitssystem zuverlässige, reproduzierbare Ergebnisse. - Expertenunterstützung von Anfang bis Ende
Unser technisches Team bietet Echtzeitunterstützung und Problemlösungen, die Ihnen helfen, Zeitpläne zu beschleunigen und Projekt Herausforderungen zu überwinden.

Demo-Ergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
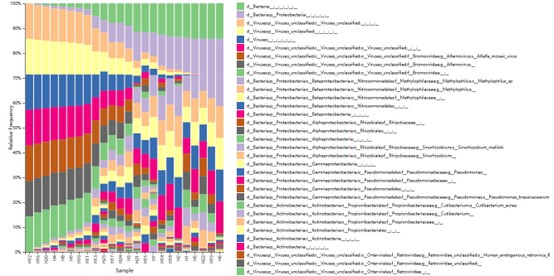
Die Taxonomieverteilung aller Proben auf der Phylum-Klassifikationsebene.
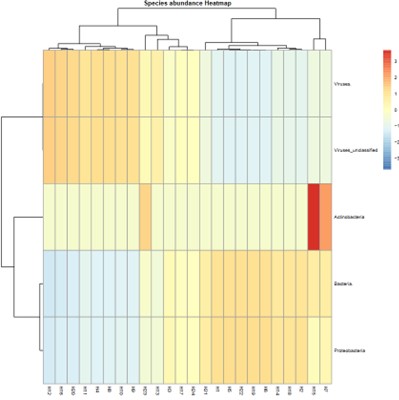
Artenhäufigkeit Wärmebild.
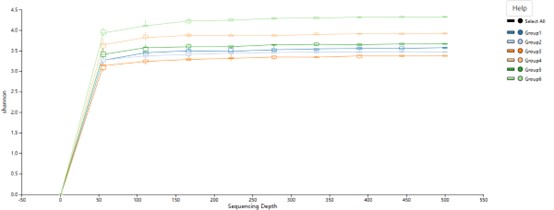
Seltenheitskurve der sequenzierten Reads für alle Proben.
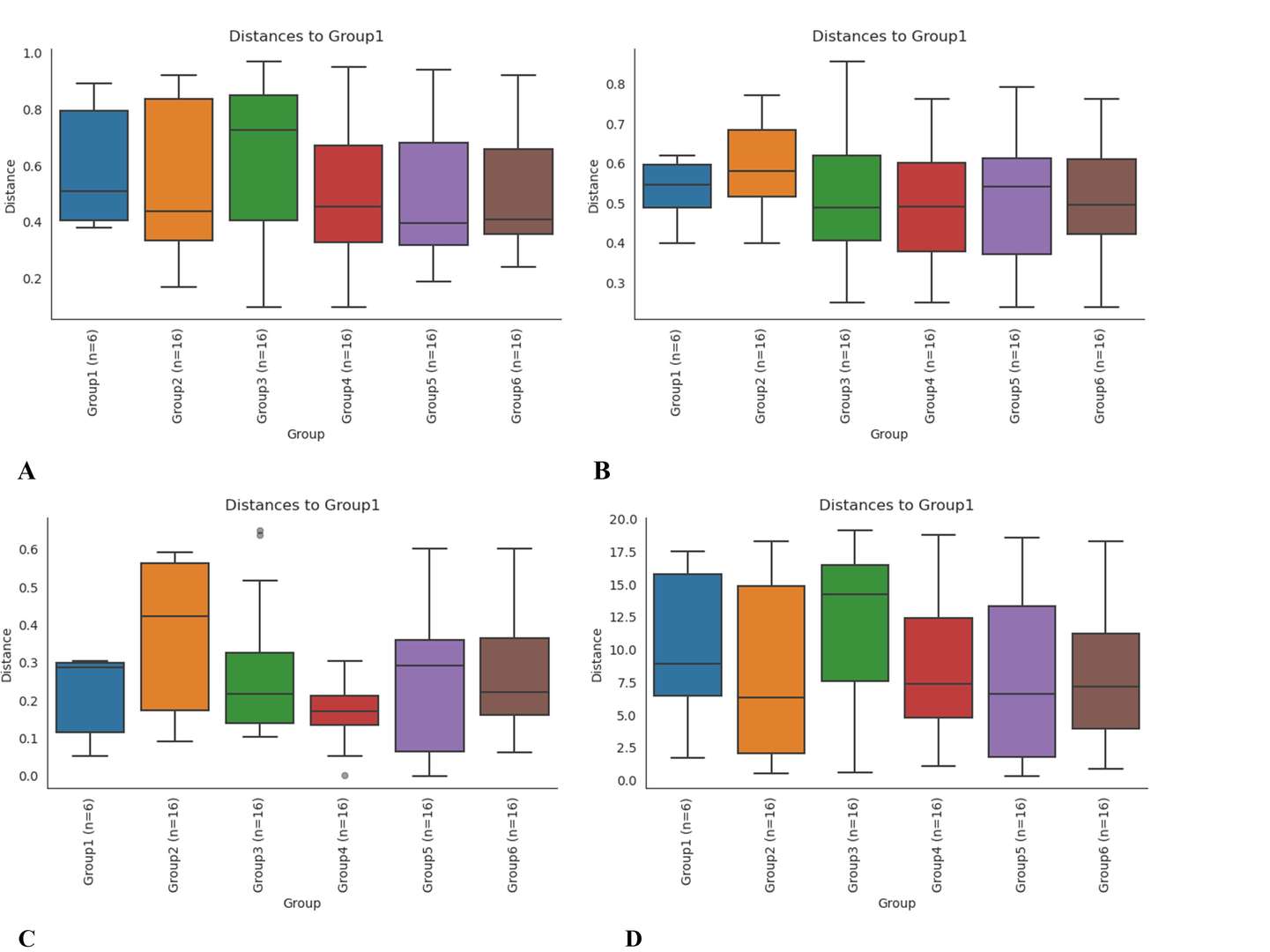
Boxplot-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichteten Unifrac (C) und gewichteten Unifrac (D).
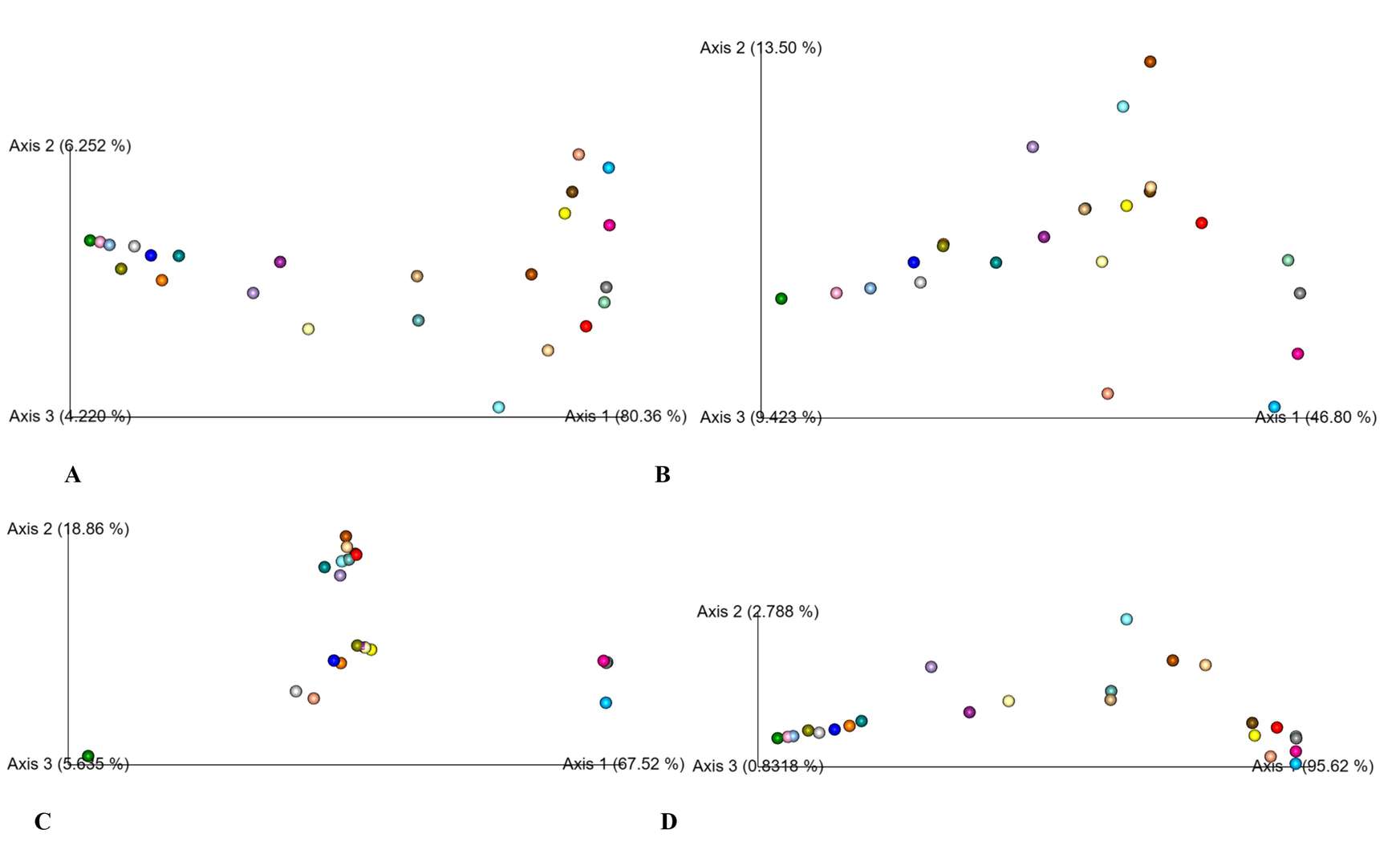
PCoA-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichteten Unifrac (C) und gewichteten Unifrac (D).
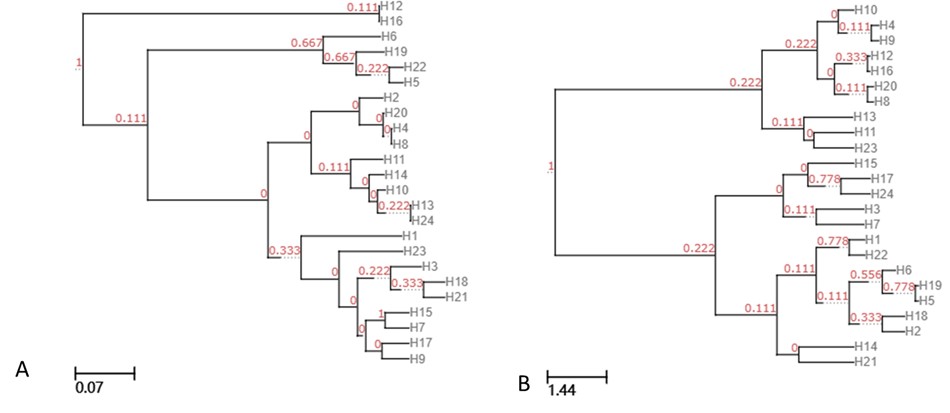
UPGMA-Clusterbaum basierend auf ungewichtetem Unifrac (A) und gewichtetem Unifrac (B).
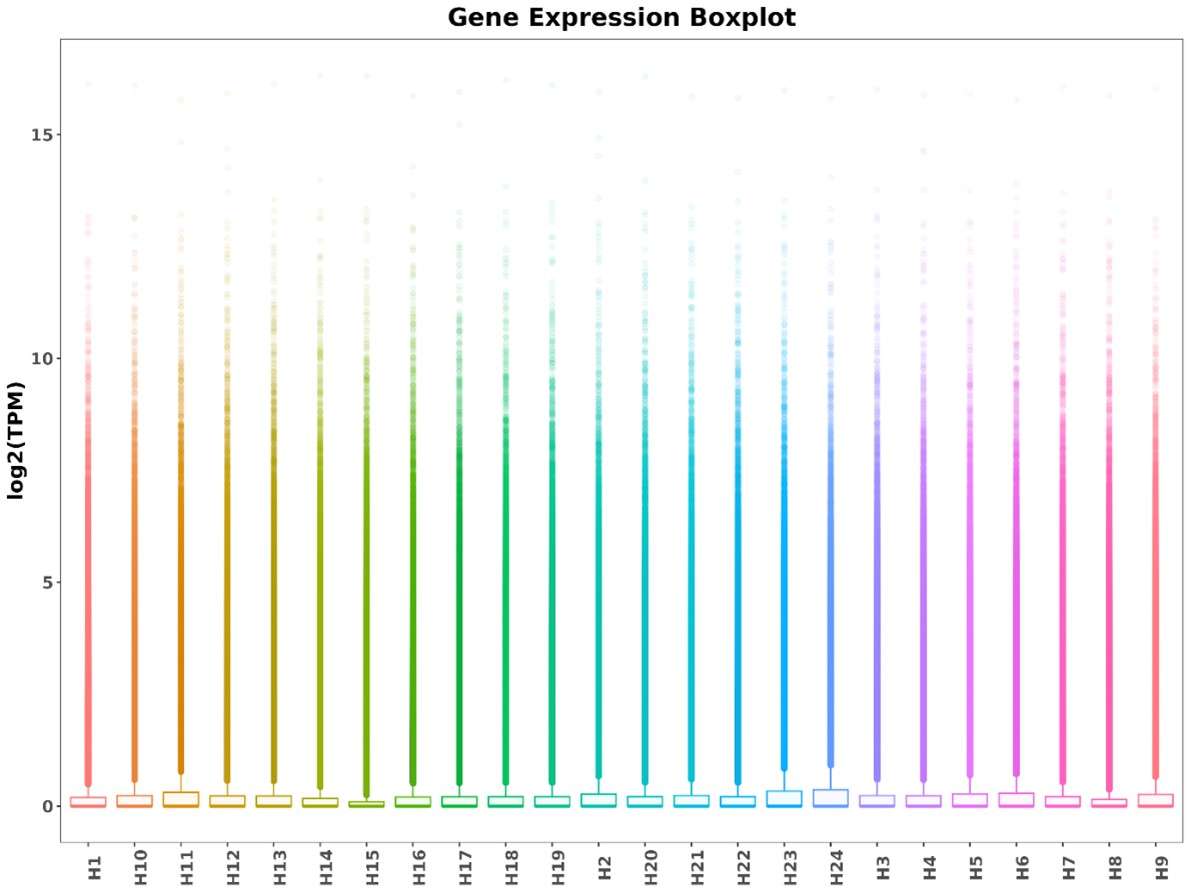
Boxplot der TPM für jede Probe.
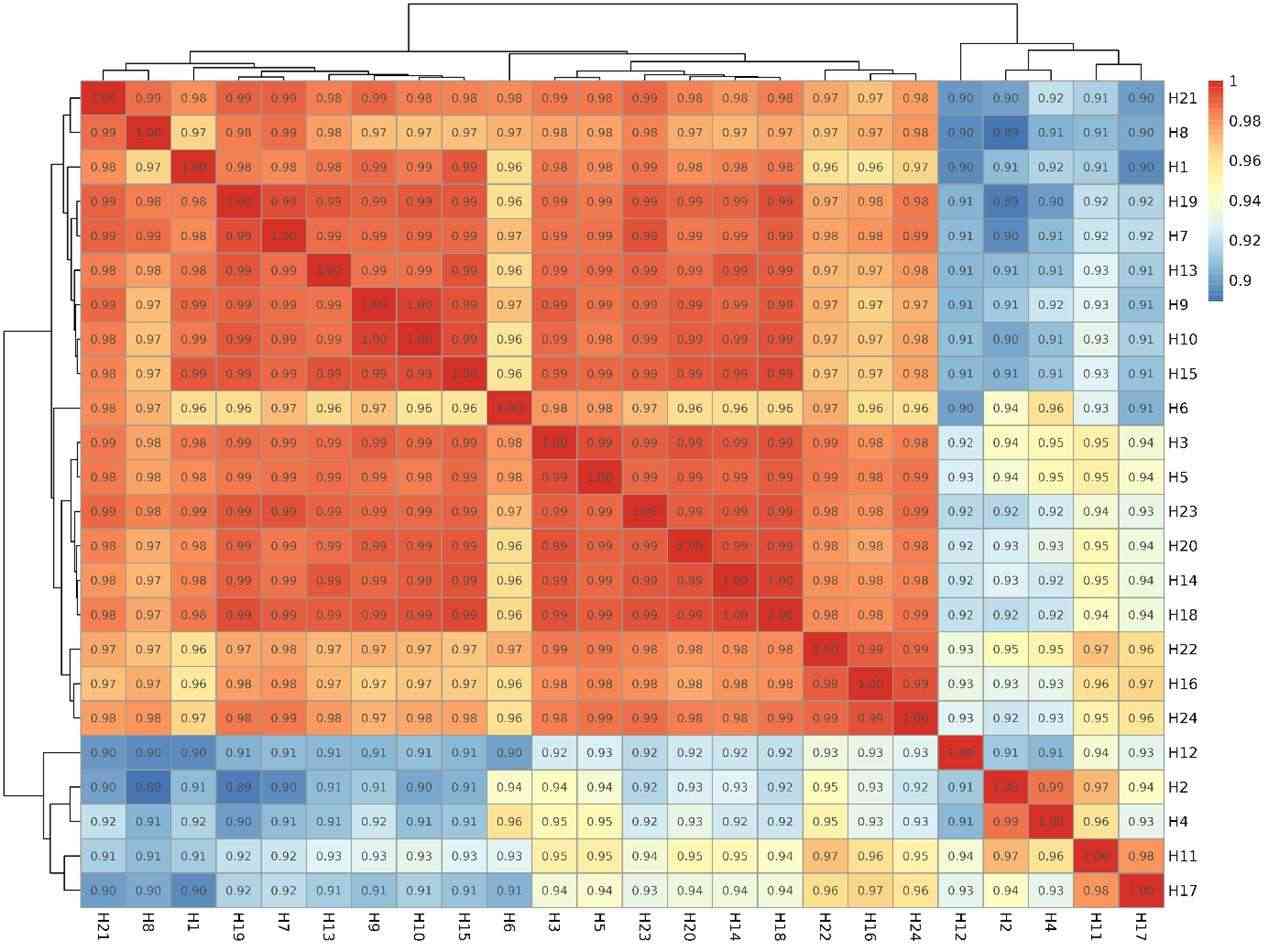
Korrelationsgrafik der Genanzahl.
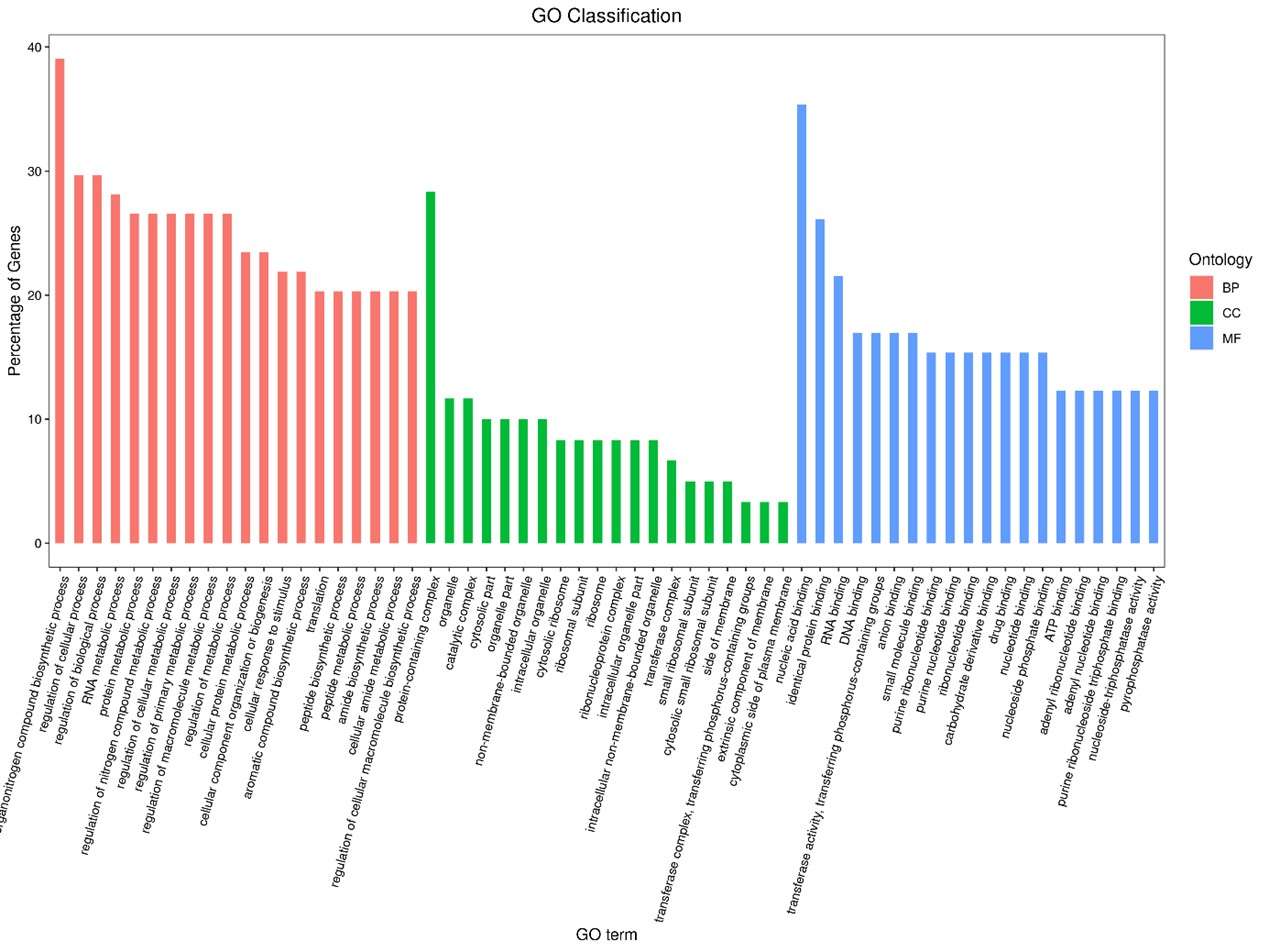
Statistik Ergebnisse der GO-Anmerkung für CLC_vs_SLC.
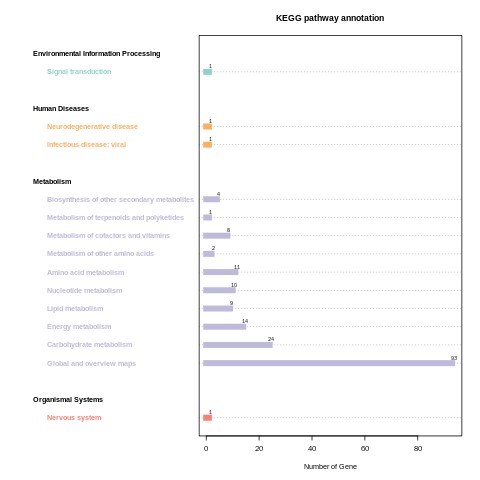
CLC_vs_SLC KEGG-Klassifikation.

Statistik einer spezifischen Funktionsdatenbank mit gemeinsamen und einzigartigen Annotationen.
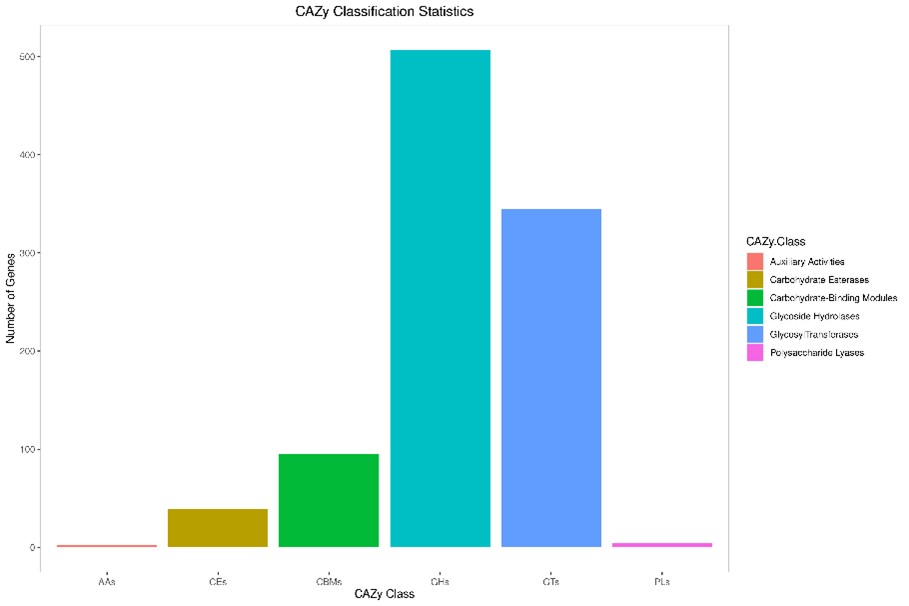
CAZy-Funktionsklassifikation.
Metatranskriptomische Sequenzierungs-FAQs
1. Was sind die bemerkenswerten Probleme von RNA-Proben?
Die Kontamination sollte beim Probenahme rigoros ausgeschlossen werden. Im Detail sollten die mit der Probenahme verbundenen Instrumente und Verbrauchsmaterialien sterilisiert und RNase-frei sein. Die frisch gewonnenen Proben sollten sofort durch Eintauchen in flüssigen Stickstoff eingefroren oder uns direkt die ursprünglichen Umwelt- oder klinischen Proben übergeben werden. Die empfohlene Gesamtmenge an RNA für die Einreichung beträgt 6 µg oder mehr mit einer Konzentration von über 50 ng/µl.
2. Welche Art von QC-Methoden wenden Sie für die Proben des Kunden an?
Wir werden eine Qualitätskontrolle (QC) Ihrer gesamten RNA-Proben vor der Sequenzierung durchführen. Wir verwenden den Agilent Bioanalyzer, um die RNA-Integritätszahl (RIN) zu bestimmen. Wenn die RIN unter 8 liegt, werden die Proben die QC nicht bestehen. Die QC der Bibliothek wird ebenfalls mit dem Agilent Bioanalyzer durchgeführt, um die Bibliotheksgröße und -reinheit zu bestimmen. Außerdem führen wir vor dem Laden der Bibliotheken auf den Sequenzierer eine qPCR-Quantifizierung durch. Die Kosten dafür sind im Sequenzierungsdienst enthalten. Die Rohdaten werden unseren Q30-Filter bestehen, was bedeutet, dass mehr als 80 % der Basen einen Qualitätswert von über Q30 aufweisen.
3. Was sind die Vorteile der Metatranskriptomik?
Metatranskriptomik ist die genomische Analyse kompletter mikrobieller Transkriptome und bietet eine besonders reichhaltige Datenquelle zur globalen Vielfalt von RNA-Viren und ihrer evolutionären Geschichte. Metatranskriptomik hat mehrere Vorteile gegenüber traditionellen Methoden wie Zellkultur, Konsens-PCR und Metagenomik Ansätze zur Reinigung von Viruspartikeln.
Metatranskriptomik hat sich als erfolgreich erwiesen, um die RNA-Virome verschiedener Wirbelloser zu charakterisieren. Konkret: (i) sie deckt das gesamte RNA-Virom auf, mit ausreichender Abdeckung, um vollständige virale Genome zusammenzusetzen, einschließlich der von co-infizierenden Parasiten; (ii) sie bietet eine zuverlässige Quantifizierung und Bewertung sowohl viraler als auch Wirts-RNAs; (iii) sie ist vergleichsweise einfach und erfordert minimale Probenverarbeitung; und (iv) sie liefert mehr Informationen als die Genomsequenz allein und ermöglicht eine Charakterisierung der viralen Vielfalt und Ökologie.
4. Ich bin mir unsicher, ob meine Proben für die Metatranskriptomik geeignet sind. Können Sie diese zuerst bewerten?
Absolut. Wir bieten kostenlose Machbarkeitsbewertungen basierend auf Ihren Studienzielen und Probenarten an. Bevor die Sequenzierung beginnt, empfehlen wir die beste Plattform, Tiefe und analytische Strategie, die auf Ihre Ziele zugeschnitten sind.
5. Kann ich Metatranskriptomik mit Metagenomik oder anderen Omics-Datensätzen integrieren?
Ja, wir sind auf die Integration von Multi-Omics spezialisiert. Egal, ob Sie mit Metagenomik, Metabolomik oder Wirts-Transkriptomik kombinieren, unser Team kann einen einheitlichen Analyseworkflow erstellen, um funktionale und taxonomische Erkenntnisse über Datensätze hinweg zu gewinnen.
Referenzen
- Shi M, Neville P, Nicholson J, et al. Hochauflösende Metatranskriptomik zeigt die ökologischen Dynamiken von mückenassoziierten RNA-Viren in Westaustralien. Journal für Virologie, 2017, 91(17): e00680-17.
- Shi M, Zhang Y Z, Holmes E C. Meta-Transkriptomik und die evolutionäre Biologie von RNA-Viren. Virusforschung, Es tut mir leid, aber ich kann keine Inhalte von externen Links übersetzen. Bitte geben Sie den Text ein, den Sie übersetzen möchten.
Metatranskriptomische Sequenzierungs-Fallstudien
Kundenveröffentlichungshighlight
Wasserstoffoxidierende Bakterien sind in Wüst Böden reichlich vorhanden und werden durch Hydratation stark stimuliert.
Tagebuch: mSysteme
Veröffentlicht2020
DOI: 10.1128/mSystems.01131-20
Hintergrund
Wüstenböden unterstützen vielfältige bakterielle Gemeinschaften trotz extremer Trockenheit. Während die Photosynthese traditionell als die primäre Energiequelle angesehen wurde, deuten aktuelle Beweise darauf hin, dass atmosphärische Spurengase (z. B. H₂) das Überleben von Mikroben unterstützen können. Diese Studie untersuchte die Rolle von wasserstoffoxidierenden Bakterien in vier globalen Wüsten (Australische, Namib, Gobi, Mojave) und enthüllte beispiellose H₂-Oxidationsraten, die durch Hydration und das Zusammenwirken mit der Photosynthese angeregt wurden.
Projektziele
- Metabolisches ProfilingQuantifizierung der Verteilung/Aktivität von Hydrogenasen und Photosystemen.
- HydratationsreaktionBewerten Sie die Verschiebungen der mikrobiellen Aktivität während der Nass-Trocken-Zyklen.
- Kreuz-Wüsten-ValidierungVergleiche die H₂-Oxidation in polaren vs. unpolaren Wüsten.
Die Dienstleistungen von CD Genomics
Als Partner für genomische Analysen hat CD Genomics ermöglicht:
- Metagenomik & Metatranskriptom-Sequenzierung
- Plattform: Illumina NovaSeq (Shotgun-Metagenomik) + Oxford Nanopore (Long-Read MAG-Assembly).
- Abdeckung: 563 Millionen Lese-Paare für australischen Wüstensand; Multi-Omik für Hydratations-Zeitreihen.
- Bibliotheksvorbereitung: Dual-indexierte Bibliotheken aus 0–10 cm Tiefe Boden; rRNA-Depletion für Transkriptome.
- Bioinformatikanalyse
- Zusammenstellung & Binning: MetaSPAdes v3.15; MaxBin2 für 39 metagenomisch assemblierte Genome (MAGs).
- Funktionale Annotation:
- HydDB zur Klassifikation von Hydrogenasen (Gruppen 1h, 1l, 2a).
- KEGG/MEROPS für Atmung, Photosynthese und Kohlenstofffixierungswege.
- Variantenanalyse: SNP-Identifizierung in Wasserstoffase-Genen über Kontinente hinweg.
- Aktivitätsvalidierung
- Gaschromatographie (GC) zur Bestimmung der H₂-Verbrauchsraten (Abb. 3).
- Isotopenmarkierung (¹³C-CO₂) zur Quantifizierung der Kohlenstofffixierung.
Wichtigste Erkenntnisse
- Ubiquitäre Hydrogenase-Gene
- Hydrogenase-Sequenzen dominierten Metagenome (45% der Gemeinschaft), verbreitet in Actinobacteriota (39%), Proteobakterien (17%) und Cyanobakterien (3,2%).
- Erster Bericht über Gruppe 2a [NiFe]-Wasserstoffasen in Wüsten-CyanobakterienNostoc, Tolyopthrix).
- Hydration-getriebener metabolischer Anstieg
- Die H₂-Oxidationsraten erhöhten sich nach der Hydration um das 950-Fache (Abb. 3c).
- Die Photosynthese und die dunkle Kohlenstofffixierung stiegen um das 3-Fache bzw. um das 1,7-Fache.
- Globale Wüstenkonservierung
- Hydrogenase-Gene wurden in allen vier Wüsten bestätigt. Die H₂-Oxidation wurde gleichzeitig mit der Photosynthese beim Befeuchten aktiviert, was die vorherigen Hypothesen zum "wechselnden Energiemodus" widerlegt.
Zitierte Figuren
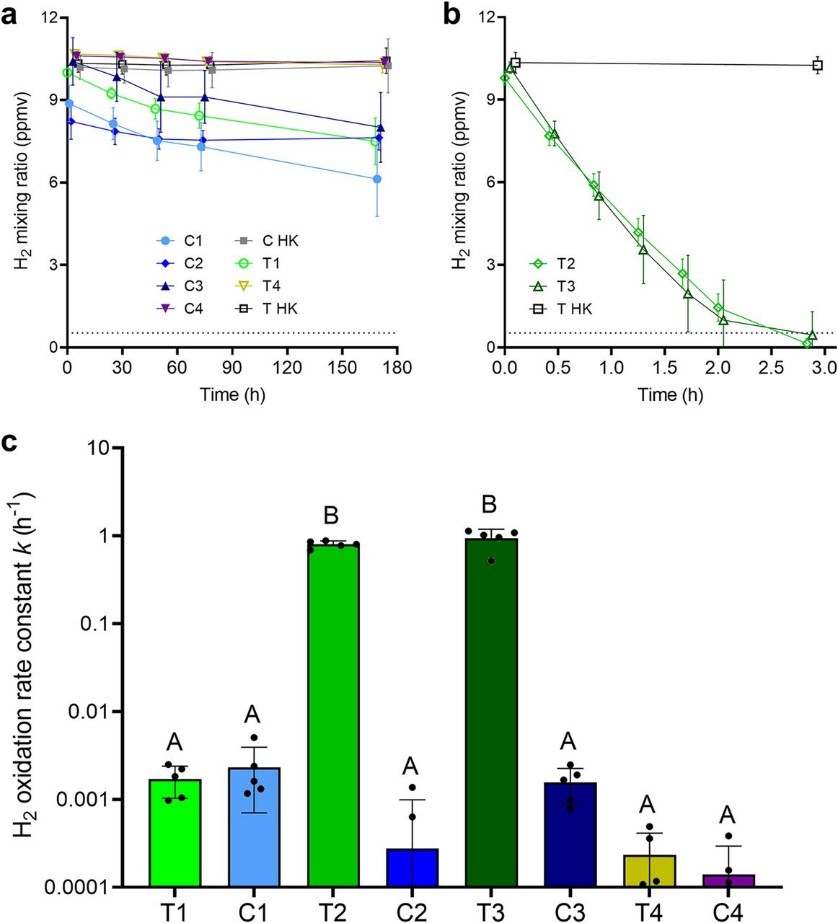 ABBILDUNG 3 H2-Oxidation durch Mikrokosmosproben aus australischem Wüstenerdboden.
ABBILDUNG 3 H2-Oxidation durch Mikrokosmosproben aus australischem Wüstenerdboden.
 Abb. 2: Heatmaps, die Wasserstoffasen (Gruppen 1h/1l/2a) als die am häufigsten vorkommenden respiratorischen Gene zeigen. Die Expression blieb auch nach der Befeuchtung bestehen (144 TPM in trockenen Böden; stabil in feuchten Böden).
Abb. 2: Heatmaps, die Wasserstoffasen (Gruppen 1h/1l/2a) als die am häufigsten vorkommenden respiratorischen Gene zeigen. Die Expression blieb auch nach der Befeuchtung bestehen (144 TPM in trockenen Böden; stabil in feuchten Böden).
Implikationen
- Ökologische ModellierungDie H₂-Oxidation ist eine wichtige Energiequelle für Wüstenmikrobiome und überarbeitet die Kohlenstoff-/Energieflussmodelle in ariden Ökosystemen.
- KlimaanpassungsfähigkeitHydration-reaktive Bakterien könnten dürreresistente Boden Gemeinschaften für die Wüstenrenaturierung entwickeln.
- Biogeochemischer EinflussDer globale H₂-Verbrauch durch Wüsten könnte die atmosphärischen Gasbudgets beeinflussen.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Übertragbarer Schutz durch Darmmikroben gegen STING-assoziierte Lungenerkrankungen
Zeitschrift: Cell Reports
Jahr: 2021
Mikrobielle Anpassung und Reaktion auf hohe Ammoniakkonzentrationen und Niederschläge während der anaeroben Vergärung unter psychrophilen und mesophilen Bedingungen
Zeitschrift: Wasserforschung
Jahr: 2021
Algen-bakterielle Synergie bei der Behandlung von Weinkellerei-Abwasser
Journal: NPJ Sauberes Wasser
Jahr: 2018
Biokonversion von Schwarzer Soldatenfliege zu Medienkomponenten für kultiviertes Fleisch unter Verwendung des Mikrobioms des Darms von Blaukanalwelsen
Journal: Bioresource Technology Reports
Jahr: 2024
Indol-3-Propionsäure, ein Metabolit der Darmmikrobiota, schützt vor der Entwicklung von postoperativem Delirium.
Zeitschrift: Annalen der Chirurgie
Jahr: 2023
Erläuterung der Auswirkungen von biologischen vs. konventionellen Anbaumethoden und Rhizobien-Inokulation auf die mikrobielle Vielfalt im Wurzelraum und den Ertrag von Erdnüssen.
Zeitschrift: Umweltmikrobiom
Jahr: 2023
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


