
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Nanopore-RNA-Methylierungs-Sequenzierungsdienst
CD Genomics bietet modernste Nanopore-RNA-Methylierungs-Sequenzierungsdienste an, um m6A-Modifikationsmuster direkt aus RNA-Molekülen zu enthüllen. Unser Hochdurchsatzverfahren ermöglicht eine Analyse mit Einzelbasenauflösung und bietet detaillierte Einblicke in die RNA-Epigenetik. Dieser Dienst unterstützt Untersuchungen zur Genregulation, RNA-Stoffwechsel und Krankheitsmechanismen.
Was ist Nanopore-RNA-Sequenzierung?
DNA-Methylierung und RNA-Methylierung sind Prozesse, die durch Methyltransferasen erleichtert werden, wobei eine Methylgruppe (CH3) an ein bestimmtes Atom eines DNA- oder RNA-Moleküls angefügt wird. In zellulärer RNA wurden über 100 Arten chemischer Modifikationen identifiziert. Verschiedene RNA-Methylierungsmodifikationen umfassen m6A RNA-Methylierung, m5C RNA-Methylierung, m1A RNA-Methylierung, m7G RNA-Methylierung und mehr. Unter diesen ist die am prominentesten und umfassendsten untersuchte die m6A RNA-Methylierung, die am sechsten Stickstoffatom des Adenin-Restes in RNA-Molekülen auftritt (N6-Methyladenosin, m6A). Diese Modifikation stellt die häufigste posttranskriptionale Modifikation in eukaryotischer mRNA dar und macht etwa 80 % aller RNA-Methylierungsmodifikationen aus.
ONT-RNA-Methylierungssequenzierung wird mit Nanopore-Technologie durchgeführt. Nanoporen sind kleine Poren mit einem Durchmesser im Nanometerbereich. Wenn ein einzelner RNA-Strang durch eine Nanopore hindurchgeht, erzeugt dies Veränderungen im elektrischen Strom. Diese Stromschwankungen können präzise aufgezeichnet und von spezifischen Algorithmen entschlüsselt werden, um die Sequenzinformationen der RNA zusammen mit ihren Methylierungsmodifikationen offenzulegen. Der Schlüsselbereich der ONT-Technologie liegt in ihrer Fähigkeit, lange RNA-Moleküle direkt zu sequenzieren und verschiedene Modifikationen an RNA zu erkennen, einschließlich N6-Methyladenin (m6A), 5-Methylcytosin (m5C) und anderen.
Methoden zur Erkennung von RNA-Methylierung
MeRip-seq (Methylierte RNA-Immunpräzipitation):
MeRip-seq basiert auf dem Prinzip der antikörperspezifischen Bindung an methylierten Basen. Es nutzt m6A-spezifische Antikörper zur Immunpräzipitation, um RNA-Fragmente anzureichern, die einer Methylierung unterliegen. Anschließend wird Hochdurchsatz-Sequenzierung eingesetzt, um Transkripte mit m6A-Modifikationen zu detektieren. Diese Methode kann jedoch nur Regionen mit hoher Methylierung identifizieren und bietet keine Einzelbasenauflösung zur Erkennung von RNA-Methylierung.
Nanopore-Sequenzierungstechnologie
Nanoporen-Sequenzierung ist ein Langzeit-Sequenzierung technologie, die auf der elektrischen Signalermittlung von Basensequenzen basiert. Verschiedene Modifikationen der RNA-Basen führen zu Variationen in der Obstruktionsgröße, während sie durch einen Nanoporenkanal passieren, was charakteristische elektrische Signale erzeugt. Die Echtzeitüberwachung dieser Signale ermöglicht die Bestimmung der entsprechenden Basentypen und ob sie Basismodifikationen tragen. Mit anderen Worten, die Nanoporen-Sequenzierung kann direkt durchführen Vollständige Transkriptom-Sequenzierung auf natürlichen RNA-Proben ohne die Notwendigkeit spezifischer Antikörperbindung. Es ermöglicht die Erkennung von m6A (N6-Methyladenosin) Methylierungsmodifikationen auf RNA mit Einzelbasenauflösung und bietet gleichzeitig quantitative Gene/transkript Ausdruck und Identifizierung der Transkriptstruktur.
Prinzipien der Nanoporen-RNA-Methylierungssequenzierung
Die Nanoporen-Sequenzierungstechnologie nutzt die Erkennung von Signalen, die erzeugt werden, wenn ein einzelnes Molekül durch eine Nanopore hindurchgeht, wodurch ein Potentialunterschied auf beiden Seiten der Pore entsteht. Der Durchmesser der Nanopore erlaubt es nur, einzelne Nukleotidpolymere hindurchzulassen. Die geladene Natur der verschiedenen Basen, einschließlich derjenigen mit methylierter Modifikation, variiert, was die Erkennung der entsprechenden Basentypen und Methylierungsinformationen durch Unterschiede in den elektrischen Signalen ermöglicht.
Vorteile des Nanopore-RNA-Methylierungs-Sequenzierungsdienstes
- Einzelne Nukleotidauflösung über das gesamte Transkriptom: Identifizierung von m6A-Methylierungsmodifikationen mit einzelner Nukleotidauflösung im gesamten Transkriptom.
- Direkte Ablesung von m6A-Methylierungsinformationen: Direkte Extraktion von m6A-Methylierungsinformationen ohne die Notwendigkeit von Immunpräzipitationsanreicherungsversuchen.
- Gleichzeitige Quantifizierung der Gen-/Transkriptexpression und Transkriptstruktur: Durchführung sowohl der quantitativen Analyse der Gen-/Transkriptexpression als auch der Untersuchung der Transkriptstruktur gleichzeitig.
- Fehlen von reverser Transkription und PCR-Bias: Nanopore-Direkt-RNA-Sequenzierung eliminieren die Notwendigkeit von Störungen, reverser Transkription und PCR-Amplifikation, was eine direkte ermöglicht Vollständige Transkriptom-Sequenzierung von natürlichen RNA-Proben.
Anwendungen der Nanoporen-RNA-Methylierungssequenzierung
- Untersuchung der funktionalen Rolle molekularer Teilnehmer, die an RNA-Methylierungsprozessen während Krankheiten oder spezifischer Lebensprozesse beteiligt sind.
- Entschlüsselung der Veränderungen in RNA-Methylierungsmodifikationen während Krankheiten oder bestimmter Lebensprozesse, wodurch krankheitsspezifische m6A RNA-Methylierungsmuster aufgeklärt werden.
- Untersuchung der Beziehung zwischen spezifischen RNA-Methylierungsmodifikationen und Krankheiten während von Krankheiten oder spezifischen Lebensprozessen.
- Identifizierung neuer Moleküle, die an RNA-Methylierungsmodifikationen beteiligt sind, oder Etablierung neuer Methoden zur Untersuchung von RNA-Methylierungsmodifikationen.
Nanopore-RNA-Methylierungs-Sequenzierungs-Workflow
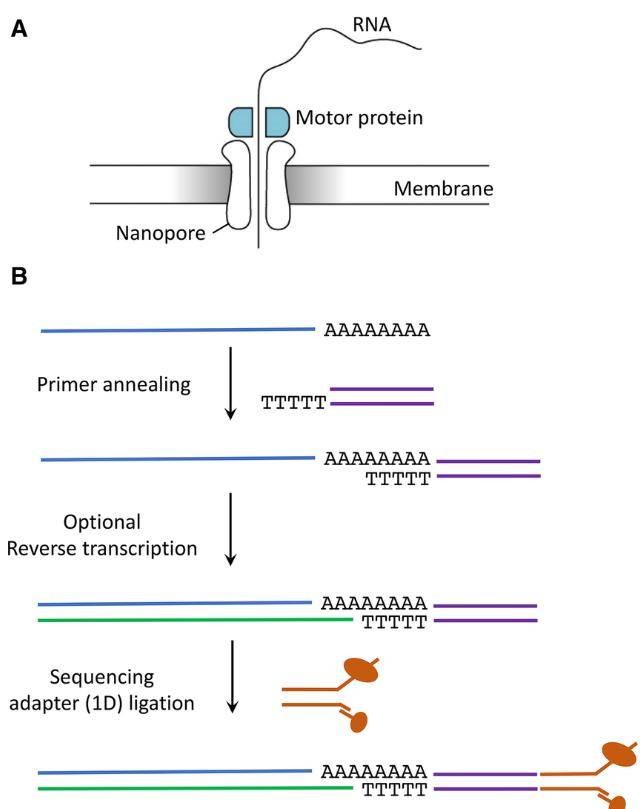 Abb. 1. Schritte zur Bibliotheksvorbereitung mit Oxford Nanopore Technologies. (Nicky Jonkhout et al., 2017)
Abb. 1. Schritte zur Bibliotheksvorbereitung mit Oxford Nanopore Technologies. (Nicky Jonkhout et al., 2017)
Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
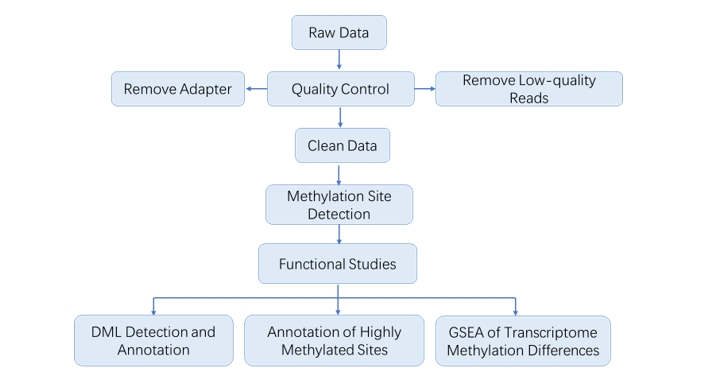
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an: m6A RNA-Methylierungserkennung:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Nanopore-RNA-Methylierungssequenzierung für Ihre Schreibanpassung.
Erforschen Sie RNA-Methylierung mit der Nanopore-RNA-Methylierungssequenzierung von CD Genomics. Wir bieten direkte RNA-Sequenzierung und umfassende bioinformatische Analysen, um epigenetische Muster aufzudecken. Kontaktieren Sie uns, um Ihre Forschungserkenntnisse voranzubringen.
Referenz
- Jonkhout N, Tran J, Smith MA, et al. Die RNA-Modifikationslandschaft bei menschlichen Krankheiten. RNA2017, 23(12):1754-69.
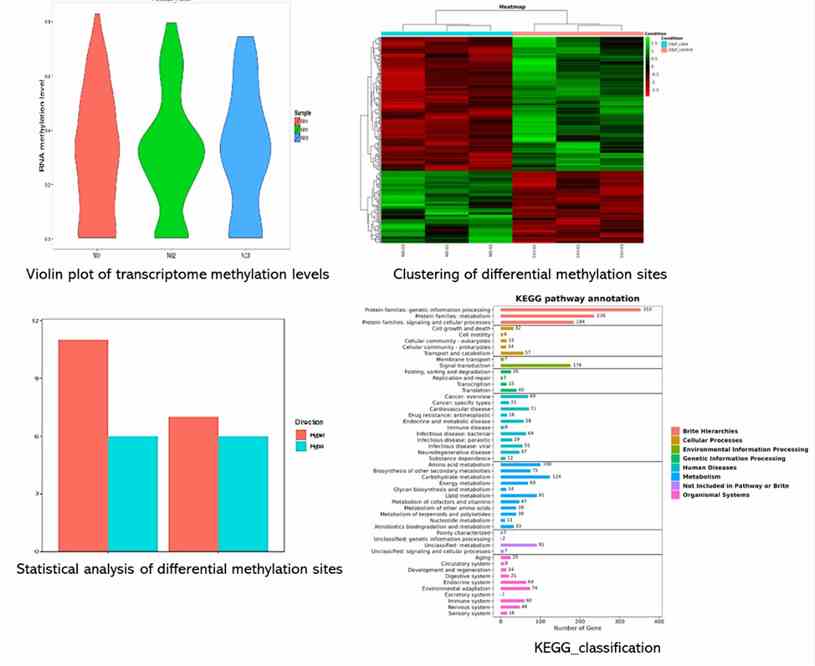
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
Nanopore RNA-Methylierung Seq FAQs
1. Welche Moleküle sind an RNA-Modifikationsprozessen beteiligt?
Im Prozess der RNA-Modifikation sind mehrere Schlüssel-Moleküle beteiligt. Writer, wie METL3 und METTL14, katalysieren die Methylierung von RNA, wobei WTAP eine entscheidende Rolle in diesem Methyltransferase-Komplex spielt. Diese Enzyme erleichtern die m6A-Methylierung von mRNA (und anderen nukleären RNAs) sowohl in vitro als auch in vivo. Auf der anderen Seite sind Eraser wie FTO und ALKBH5 dafür verantwortlich, m6A-Methylierungssignale von RNA zu entfernen und somit den Demethylierungsprozess zu vermitteln. Reader hingegen sind Proteine, die die modifizierten Informationen in RNA interpretieren und an nachgelagerten Prozessen wie Translation und Spleißen teilnehmen. Proteine mit YTHDF-Domänen können beispielsweise m6A in mRNA erkennen und binden, was zu einer Verringerung der mRNA-Stabilität führt und deren Abbau fördert.
2. Welche Arten von RNA-Methylierung können mit der Nanopore-RNA-Methylierungssequenzierung nachgewiesen werden?
Dieses Verfahren kann verschiedene RNA-Methylierungsmodifikationen nachweisen, einschließlich N6-Methyladenosin (m6A), 5-Methylcytosin (m5C) und andere.
3. Welche Forschungsanwendungen können von der Nanopore-RNA-Methylierungssequenzierung profitieren?
Diese Technologie ist wertvoll für das Studium der epigenetischen Regulation der Genexpression, der RNA-Verarbeitung und verschiedener biologischer Prozesse, die von RNA-Modifikationen beeinflusst werden. Sie ermöglicht es Forschern, dynamische Veränderungen in den RNA-Methylierungsmustern unter verschiedenen Bedingungen zu untersuchen.
4. Wie vergleicht sich die Nanopore-RNA-Methylierungssequenzierung mit anderen Methoden wie MeRIP-seq oder CM-seq?
Im Gegensatz zu antikörperbasierten Methoden (z. B., MeRIP-seq) oder chemische Modifikationsansätze (z. B. CM-seq) liest die Nanopore-RNA-Methylierungssequenzierung RNA-Sequenzen und Modifikationen in Echtzeit und bietet Vorteile in Bezug auf umfassende Abdeckung und reduzierte Verzerrung.
5. Was sind einige typische Ergebnisse der Nanopore-RNA-Methylierungssequenzierung?
Die Ergebnisse umfassen detaillierte Methylierungsprofile von RNA-Transkripten, die Visualisierung von Methylierungsmustern (z. B. Violin-Plots), die Analyse unterschiedlicher Methylierung sowie funktionale Annotationen von methylisierten Stellen im Transkriptom.
Nanopore-RNA-Methylierungs-Sequenzierungs-Fallstudien
FIONA1-vermitteltes m6Eine Modifikation reguliert den floralen Übergang in Arabidopsis
Journal: Fortschrittliche Wissenschaft
Impactfaktor: 17,521
Veröffentlicht: 05. Januar 2022
Hintergrund
m6A ist eine häufige mRNA-Modifikation, die für die Genregulation entscheidend ist und die mRNA-Verarbeitung, Stabilität und Translation beeinflusst. Techniken wie m6A-seq, m6A-CLIP und Nanoporen-Sequenzierung ermöglichen eine präzise Kartierung von m6A-Stellen im gesamten Transkriptom. In ArabidopsisDie Ablagerung von m6A umfasst komplexe Methyltransferasen, die spezifische mRNA-Regionen und -Motive anvisieren. FIO1, das als neuartiger m6A-Methyltransferase identifiziert wurde, beeinflusst die Blütezeit, indem es die m6A-Spiegel in Schlüsselengenen wie SOC1 reguliert. Die Nanopore-Sequenzierung liefert Einblicke in die Dynamik von m6A und hebt die Rolle von FIO1 in der Pflanzenentwicklung und der Kontrolle der Genexpression hervor.
Materialien & Methoden
Probenvorbereitung
- Arabidopsis (Ein. Thaliana)
- Sechs Tage alte Sämlinge
- Plasmidkonstruktion
- Gesamt-RNA-Extraktion
Sequenzierung
- m6A-Modifikationsstellenanalyse
- Differenzielle Expressionsanalyse
- Analyse des alternativen Spleißens
- Ausdrucksanalyse
- Statistische Analyse
Ergebnisse
Die Störung von FIO1 in Arabidopsis reduziert die globalen mRNA m6A-Spiegel leicht. FIO1, ähnlich wie das menschliche METTL16, enthält eine konservierte Methyltransferase-Domäne und wird breit exprimiert in Arabidopsis Gewebe. Mutanten (fio1-1 und fio1-2) zeigen eine frühe Blüte, wobei fio1-2 eine verringerte FIO1-Expression aufweist. Die Analyse bestätigt einen Rückgang von etwa 14 % und 10 % der m6A-Spiegel in der Gesamt-RNA und mRNA der fio1-2-Mutanten im Vergleich zu Wildtyp-Pflanzen, was auf die Beteiligung von FIO1 an der m6A-Methylierung hinweist.
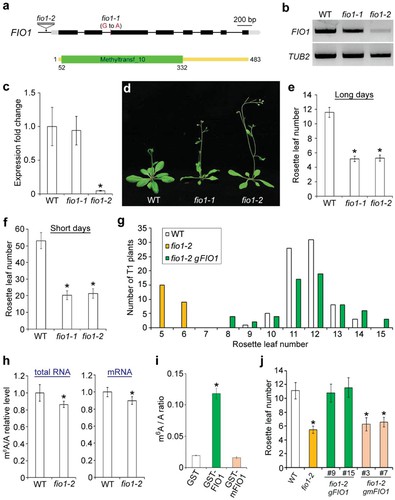 Abbildung 1 FIO1 beeinflusst die Blüte und die mRNA m6A-Spiegel in Arabidopsis.
Abbildung 1 FIO1 beeinflusst die Blüte und die mRNA m6A-Spiegel in Arabidopsis.
Nanopore-Direkt-RNA-Sequenzierung von fio1-Mutanten identifizierte 3459 hypomethylierte m6A-Stellen in kodierenden Sequenzen (CDS). Dieser Ansatz, der die xPore-Analyse verwendete, hob eine Präferenz für das YHm6AGA-Motiv hervor und zeigte Reduktionen der m6A-Spiegel hauptsächlich vor Stoppcodons. Funktionale Analysen verbanden diese Veränderungen mit der frühen Blüte in fio1-Mutanten und bestätigten die Rolle von FIO1 als m6A-Methyltransferase, die für die mRNA-Modifikation und die Entwicklungsregulation unerlässlich ist. Arabidopsis.
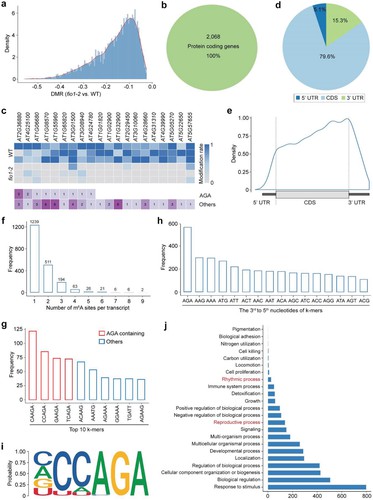 Abbildung 2 Verteilung der hypomethylierten Stellen in fio1-2.
Abbildung 2 Verteilung der hypomethylierten Stellen in fio1-2.
FIO1-vermittelte m6A-Methylierung in Arabidopsis beeinflusst die Transkript-Abundanz und alternative Polyadenylierung (APA), hauptsächlich bei Genen, die in kodierenden Sequenzen (CDS) angereichert sind. Hypomethylierte Stellen in fio1-Mutanten korrelieren mit herabregulierter Genexpression, einschließlich des wichtigen Blühregulators SOC1. FIO1 moduliert direkt SOC1 und andere Gene der circadianen Uhr, verlängert deren Expressionsperiode und beeinflusst indirekt die Blütezeit. Diese Ergebnisse heben FIO1 als eine distinct m6A-Methyltransferase hervor, die spezifische mRNA-Regionen anvisiert, um Entwicklungsprozesse in Pflanzen zu regulieren.
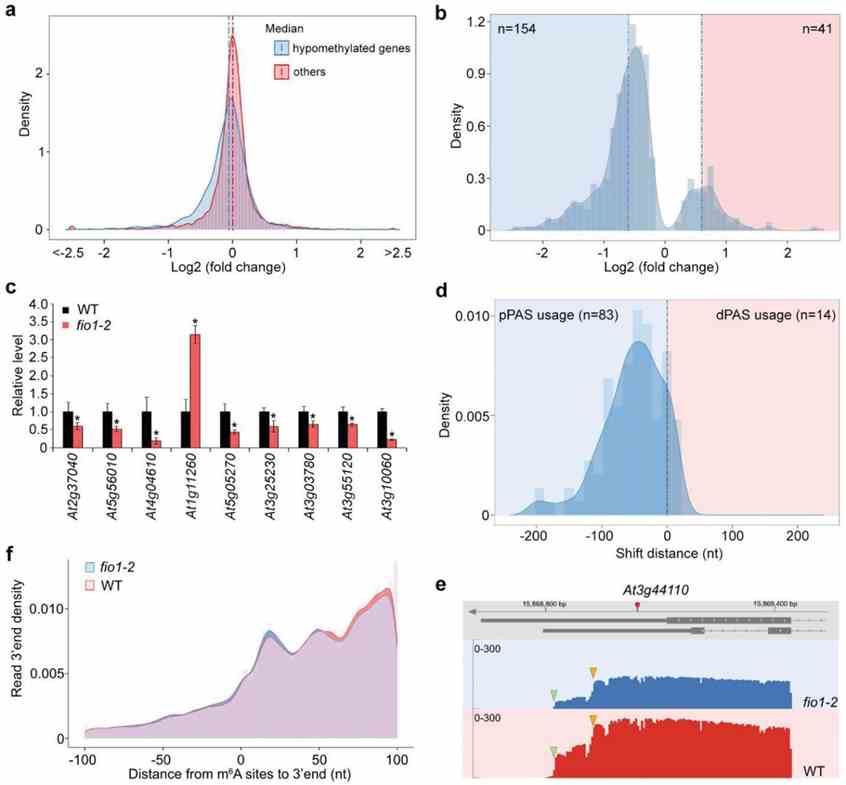 Abbildung 3 FIO1 reguliert die Transkriptmenge und alternative Polyadenylierung.
Abbildung 3 FIO1 reguliert die Transkriptmenge und alternative Polyadenylierung.
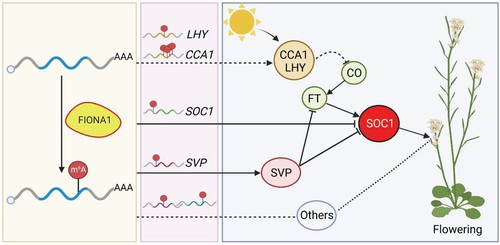 Abbildung 4 Ein vorgeschlagenes Modell, das die Funktion von FIO1 bei der m6A-Modifikation und der Kontrolle der Blühzeit darstellt in Arabidopsis.
Abbildung 4 Ein vorgeschlagenes Modell, das die Funktion von FIO1 bei der m6A-Modifikation und der Kontrolle der Blühzeit darstellt in Arabidopsis.
Fazit
Diese Studie zeigt FIO1 als eine einzigartige m6A-Methyltransferase in Pflanzen, die hauptsächlich die kodierenden Sequenzen spezifischer protein-codierender Transkripte modifiziert. Die von FIO1 vermittelte m6A-Methylierung beeinflusst die Transkriptmenge und die alternative Polyadenylierung, während sie minimale Auswirkungen auf das alternative Spleißen hat. Wichtig ist, dass FIO1 SOC1 und seine upstream-Regulatoren reguliert und somit die Blütezeit moduliert. Diese Ergebnisse erweitern unser Verständnis der m6A-Dynamik in Pflanzen und ziehen Parallelen zur von METTL16 vermittelten RNA-Modifikation bei Tieren.
Referenz
- Xu T, Wu X, Wong CE, et al. FIONA1-vermitteltes m6Eine Modifikation reguliert den Blütenübergang in Arabidopsis. Fortgeschrittene Wissenschaft2022, 9(6):2103628.
Verwandte Publikationen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die Spaltung von Phagen-DNA durch Restriktionsendonukleasen ermöglicht die Wiederbelebung aus der durch Cas13 induzierten bakteriellen Dormanz.
Zeitschrift: Nature Mikrobiologie
Jahr: 2023
IL-4 fördert die Erschöpfung von CD8.+ CART-Zellen
Zeitschrift: Nature Communications
Jahr: 2024
Fettreiche Diäten während der Schwangerschaft führen zu Veränderungen der DNA-Methylierung und Proteinexpression im Pankreasgewebe des Nachwuchses: Ein Multi-Omics-Ansatz
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2024
KMT2A assoziiert mit dem PHF5A-PHF14-HMG20A-RAI1 Subkomplex in Stammzellen des Pankreaskarzinoms und reguliert epigenetisch deren Eigenschaften.
Zeitschrift: Nature Communications
Jahr: 2023
Krebsassoziierte DNA-Hypermethylierung von Polycomb-Zielen erfordert die doppelte Erkennung von Histon H2AK119-Ubiquitinierung und der sauren Tasche des Nucleosoms durch DNMT3A.
Journal: Wissenschaftliche Fortschritte
Jahr: 2024
Genomisches Imprinting-ähnliches monoalleles väterliches Ausdrucksmuster bestimmt das Geschlecht von Kanalkatzenfischen.
Journal: Wissenschaftliche Fortschritte
Jahr: 2022
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


