
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Menschliches gesamtes Genom PacBio SMRT-Sequenzierung
CD Genomics bietet seit einigen Jahren einen genauen und erschwinglichen Dienst zur Neusequenzierung des menschlichen Genoms an. CD Genomics führt zuvor verborgene PacBio SMRT-Technologie das großes Anwendungspotenzial in der Neusequenzierung des menschlichen Genoms hat. Die langen Einzelmolekül-Lesungen zeigen strukturelle Varianten und liefern direkte Informationen zur Variantenphasing über Haplotypblöcke und Methylierung. Dies ist sehr hilfreich, um die Nützlichkeit von Präzisionsmedizin-Initiativen zur Verbesserung der menschlichen Gesundheit zu erweitern und die Entwicklung von menschlichen Ein-Gene-Krankheiten, komplexen Krankheiten und Tumorgenomen erheblich voranzutreiben.
Die Einführung der PacBio SMRT-Sequenzierung des gesamten menschlichen Genoms
eine umfassende Karte der menschlichen genomischen Variationen von größter Bedeutung für ein tiefgehendes Verständnis genetischer Merkmale und zur Unterstützung genauer Krankheitsforschung ist. Obwohl die Kurzlesesequenzierung kleine Variationen wie SNPs (Einzelnukleotid-Polymorphismen) und InDels (Einfügungen und Löschungen) zuverlässig erkennen kann, zeigt sie eine begrenzte Sensitivität bei der Identifizierung von CNVs (Kopienzahlvariationen) und SVs (strukturellen Variationen). In den letzten Jahren, Langzeit-Sequenzierung hat in der humangenomischen Forschung weit verbreitete Anwendung gefunden. Diese Technik entschlüsselt effektiv komplexe genomische Strukturen, einschließlich hochrepetitiver Genregionen und struktureller Genomvariationen, was eine umfassendere genomische Perspektive im Streben nach krankheitsassoziierten Variationen bietet. Die hohe Genauigkeit von Langzeit-Sequenzierung ermöglicht es, seltene Variationen zu entdecken, die bei der Kurzlesesequenzierung möglicherweise übersehen werden, und liefert somit genauere Informationen über genetische Variationen, die eine wesentliche Grundlage für die Präzisionsmedizin und deren grundlegende Forschung darstellen.
Die menschliche gesamte Genom-PacBio-SMRT-Sequenzierung (Single Molecule, Real-Time) von Pacific Biosciences ist eine fortschrittliche Technologie zur genomischen Analyse, die die proprietäre SMRT-Sequenzierung der Ansatz, der von Pacific Biosciences entwickelt wurde. Diese Technologie zeichnet sich durch ihre Fähigkeit aus, verlängerte Leselängen zu erzeugen, eine hohe Sequenzgenauigkeit zu demonstrieren und eine direkte Erkennung epigenetischer Modifikationen zu bieten, wodurch eine umfassende und tiefgehende Analyse des menschlichen Genoms ermöglicht wird. Bemerkenswerterweise, PacBio SMRT-Sequenzierung ist in der Lage, außergewöhnlich lange Reads zu erzeugen, die typischerweise von 10 bis 15 Kilobasenpaaren (kb) reichen und in einigen Fällen sogar diese Länge überschreiten. Dieses Merkmal erweist sich als besonders vorteilhaft für das Entschlüsseln komplexer genomischer Strukturen, wie zum Beispiel Regionen, die durch einen hohen Grad an Sequenzwiederholung oder struktureller Variation gekennzeichnet sind.
CD Genomics beschäftigt jetzt PacBio SMRT-Sequenzierung zur Durchführung umfassender Analysen menschlicher Gesamtenome. Mit der Fähigkeit, durchzuführen Whole-Genome-Sequenzierung Über verschiedene individuelle und populationbasierte Proben hinweg, gefolgt von tiefgreifenden bioinformatischen Analysen auf beiden Ebenen, ebnet diese Methode den Weg für die umfassende Erforschung genomischer Variationen. Zu diesen Variationen gehören Einzelne Nukleotidpolymorphismen (SNPs), Insertionen und Deletionen (InDels), Kopienzahlvariationen (CNVs) und strukturelle Variationen (SVs). Die intimen genomischen Einblicke, die aus dieser direkten und umfassenden Erkundung gewonnen werden, sind unverzichtbar für die Identifizierung sowohl pathogener als auch Suszeptibilitätsgene sowie für das Verständnis der Mechanismen, die dem Ausbruch von Krankheiten und den Vererbungsmustern zugrunde liegen.
Vorteile der PacBio SMRT-Sequenzierung des gesamten menschlichen Genoms
- Langzeit-Lesungen. Sie sind vorteilhaft für die Variationsinformationsermittlung des gesamten Genoms, die genaue Analyse von chromosomalen strukturellen Varianten (SVs) und Fusionsgenen.
- Keine PCR-Amplifikation. Effektiv Vermeidung des Amplifikationsbias und einfache Überbrückung von Regionen mit hohem GC-Gehalt und hoher Sequenzwiederholung, um die Integrität und Homogenität der Genomabdeckung sicherzustellen.
- Erfasst direkt epigenetische Modifikationen durch Messung der kinetischen Variation während der Baseneinfügung.
Anwendung der PacBio SMRT-Sequenzierung des menschlichen gesamten Genoms
1. SV-Erkennung
SVs repräsentieren genomische Umstellungen (typischerweise definiert als länger als 50 bp), und SVs können eine wichtige Rolle bei menschlichen Krankheiten, Evolution und genetischer Vielfalt spielen. In den letzten Jahren wurden viele erbliche Krankheiten und Krebserkrankungen mit einer großen Anzahl von SVs in Verbindung gebracht. In den letzten 25 Jahren gab es enorme Fortschritte bei der Erkennung von einzel-nukleotidvarianten (SNVs), aber intermediär große (50 bp bis 50 kb) strukturelle Varianten (SV) bleiben eine Herausforderung bei der Analyse mit Kurzleseverfahren. DNA-SequenzierungDie längste Leselänge der PacBio SMRT-Sequenzen beträgt 40~70 K, was es ermöglicht, Bereiche mit hoher Wiederholung und hoher Heterozygotie leicht abzudecken und somit die Möglichkeit zur Erkennung von SVs zu bieten.
2. Haplotyp-Genotypisierung
Ein Haplotyp (haploider Genotyp) ist eine Gruppe von Allelen in einem Organismus, die zusammen von einem einzelnen Elternteil vererbt werden. Der korrekte Genotypisierung Das Haplotype-Gen ist wichtig; zum Beispiel wird das Ergebnis einer Knochenmarktransplantation von einem nicht verwandten Spender durch die Übereinstimmung von Spender und Empfänger für HLA beeinflusst. PacBio-Langlese kann mehrere Einzel-Nukleotide und strukturelle Varianten abdecken, wodurch die Varianten direkt in Haplotypen phasiert werden.
3. DNA-Methylierung
DNA-Methylierung ist ein Prozess, bei dem Methylgruppen an das DNA-Molekül angefügt werden. Sie ist für die normale Entwicklung unerlässlich und steht im Zusammenhang mit einer Reihe von Schlüsselprozessen, einschließlich genomischer Prägung, Inaktivierung des X-Chromosoms, Repression von transponierbaren Elementen, Alterung und Karzinogenese. Es gibt viele Möglichkeiten, DNA-Methylierung zu erkennen, einschließlich Whole-Genome-Bisulfid-Sequenzierung (WGBS), Methylierte DNA-Immunpräzipitation-Sequenzierung (MeDIP) und so weiter. Aber diese Methoden sind experimentell schwer zu handhaben.
Während die PacBio-Plattform die direkte Detektion von DNA-Methylierung ohne Bisulfit-Konversion anhand der Unterschiede im Intervall der Fluoreszenzimpuls-Signale beschreiben kann. Darüber hinaus die Long-Reads-Sequenzierung ermöglicht eine gründlichere Bewertung der regionalen CpG-Methylierung und erhöht die Kapazität zur Untersuchung der Beziehung zwischen phasierten einzelsträngigen Nukleotidvarianten und allelspezifischer CpG-Methylierung.
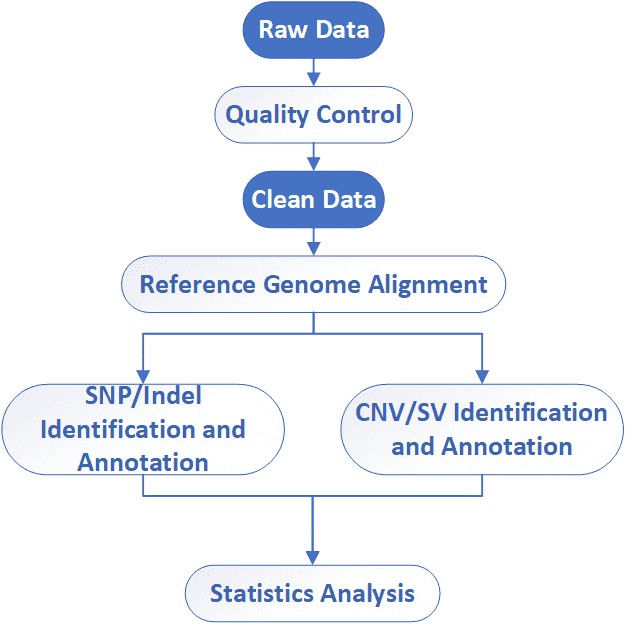
Menschliches Ganzgenom PacBio SMRT Sequenzierungs-Workflow

Dienstspezifikation
Beispielanforderungen
|
|
 |
Sequenzierungsstrategie
|
 |
Bioinformatische Analyse
CD Genomics bietet statistische und bioinformatische Datenanalysedienste für:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur menschlichen gesamten Genom-PacBio-SMRT-Sequenzierung für Ihre Schreibanpassungen.
CD Genomics bietet ein umfassendes Paket für die vollständige Neusequenzierung des Genoms an, das die Standardisierung von Proben, den Bau von Bibliotheken, tiefes Sequenzieren, die Qualitätskontrolle der Rohdaten und Bioinformatik Analyse. Wir können diese Pipeline auf Ihr Forschungsinteresse zuschneiden. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
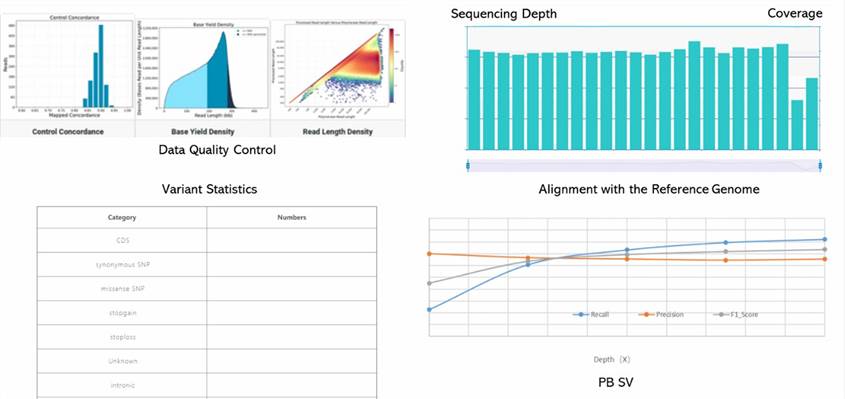
Demo-Ergebnisse
Häufig gestellte Fragen zur PacBio SMRT-Sequenzierung des menschlichen gesamten Genoms
1. Wie tief ist die resequenzierung des gesamten menschlichen Genoms?
Die Sequenzierungstiefe wird durch den Forschungszweck, die Anzahl der Proben und Ihre Bedürfnisse bestimmt. Die allgemeine Tiefe der menschlichen Whole-Genome-Resequenzierung beträgt 30X. Für die Erkennung von Keimbahnvariationen empfehlen wir eine Sequenzierungstiefe von 30-50X, wie zum Beispiel bei Forschungen zu Ein-Gen-Erkrankungen. Bei Bevölkerungsstudien mit mehreren Proben ist eine Sequenzierungstiefe von 10X ausreichend, wenn der Fokus auf SNPs liegt. Wenn Sie sich auf strukturelle Variationen in Tumorgeweben konzentrieren, empfehlen wir eine Sequenzierungstiefe von mehr als 50X.
2. Welche Methoden können verwendet werden, um die Ergebnisse zu validieren?
Whole-Genome-Resequenzierung kann verschiedene Arten von genetischen Variationen erkennen, einschließlich SNP, InDel, SV und CNV.
- PCR-Amplifikation und Sequenzierung oder SNP-Genotypisierung kann verwendet werden, um SNPs zu validieren.
- PCR-Amplifikation und Sanger-Sequenzierung kann verwendet werden, um kurze Fragmente von InDels zu validieren.
- Die Echtzeit-PCR ist nützlich zur Validierung von CNVs.
- Kleinmaßstäbliche SVs können durch PCR-Amplifikation und Sequenzierung validiert werden, während großmaßstäbliche SVs durch mikroskopische Beobachtung, wie FISH, validiert werden müssen.
3. Was sind die Vorteile der Drittgeneration der gesamten Genomsequenzierung gegenüber der Zweitgeneration der gesamten Genomsequenzierung?
Im Gegensatz zur zweiten Generation Whole-Genome-Sequenzierung, das eine Bibliothekskonstruktion mit DNA-Fragmenten von etwa 350 Basenpaaren (bp) nutzt und einen Paired-End 150 (PE150) Sequenzierungsansatz verwendet, befasst sich die Drittgeneration der gesamten Genomsequenzierung typischerweise mit Fragmenten, die größer als 10 Kilobasen (Kb) sind, mit der Fähigkeit, bis zu mehreren Megabasen (Mb) zu erweitern. Dieses Merkmal bietet besondere Vorteile bei der Erkennung von großen segmentalen strukturellen Variationen und komplexen Regionsvariationen. Darüber hinaus macht die Drittgeneration der gesamten Genomsequenzierung die PCR-Amplifikation überflüssig, wodurch eine gleichzeitige Extraktion von Methylierungsinformationen ermöglicht wird.
4. Warum lange Lesesequenzierung zur Erkennung struktureller Variationen (SV) verwenden?
strukturelle Varianten (SV), die aus Deletionen, Insertionen, Duplikationen und Inversionen bestehen, machen den Großteil der Varianten-Basenpaare im menschlichen Genom aus. Viele Studien haben eine direkte oder indirekte Beziehung zwischen SVs, die mit der menschlichen Gesundheit in Verbindung stehen, und ihren assoziierten Phänotypen gezeigt. Folglich gehört die Identifizierung genetischer Varianten und das Verständnis ihrer funktionalen Implikationen zu den kritischsten Fragen in der menschlichen Genetikforschung. Angesichts der Tendenz von SVs, innerhalb von Wiederholungsregionen aufzutreten, und der möglichen Komplexität der intra-SV-Struktur kann die Entdeckung dieser Varianten und die Genotypisierung erhebliche Herausforderungen darstellen. Daher besteht die Notwendigkeit für Langzeit-Sequenzierungstechnologien Neue Untersuchungen zu SV-Strukturen einzuleiten und Analysen zur Erkennung von SVs in der menschlichen Bevölkerung durchzuführen, ist von größter Bedeutung für das Verständnis von Krankheiten, die mit SVs in Zusammenhang stehen.
5. Wie wird die Datenqualitätskontrolle durchgeführt?
Die Datenqualitätskontrolle umfasst mehrere Schritte, um die hohe Qualität der Sequenzierungsdaten sicherzustellen:
- Bewertung der Längenverteilung der Reads: Überprüfung der Längenverteilung der erzeugten Reads, um sicherzustellen, dass sie den Erwartungen entspricht.
- Bewertung der Fehlerquote: Bewertung der Sequenzierungsfehlerquoten mithilfe interner Kontrollsequenzen oder Referenzgenomen.
- Abdeckungsbewertung: Untersuchung der Tiefe und Gleichmäßigkeit der Genomabdeckung, um eine ausreichende Datenabdeckung sicherzustellen.
6. Wie wählt man die geeignete Sequenzierungstiefe aus?
Die Wahl der Sequenzierungstiefe hängt von den spezifischen Anforderungen und Zielen der Studie ab:
- Whole Genome Assembly: Erfordert typischerweise eine höhere Abdeckungsdichte, um die Integrität und Genauigkeit der Assemblierung sicherzustellen.
- Variantenerkennung: Eine moderate Abdeckungstiefe kann ausreichen, um SNPs und InDels zu erkennen, aber die Erkennung von CNVs und SVs könnte eine höhere Abdeckung erfordern.
- Analyse epigenetischer Modifikationen: Die Abdeckungstiefe sollte ausreichend hoch sein, um eine zuverlässige Erkennung von Modifikationssignalen zu gewährleisten.
Fallstudien zur PacBio SMRT-Sequenzierung des menschlichen gesamten Genoms
Erkennung einer langen Insertion-Variante in SAMD12 durch SMRT-Sequenzierung: Auswirkungen der Langread-Whole-Genome-Sequenzierung auf Krankheiten mit Wiederholungserweiterungen
Zeitschrift: Zeitschrift für Humangenetik
Impactfaktor: 5,881
Veröffentlicht: 17. Dezember 2018
Hintergrund
Kurzlese Next-Generation-Sequenzierung (NGS) wird häufig in der medizinischen Forschung und genetischen Tests verwendet, um pathogene Einzel-Nukleotid-Varianten und kleine Einfügungen und Löschungen (Indels) nachzuweisen. Diese Technologie kann jedoch strukturelle Variationen (SVs) über Hunderte bis Zehntausende von Basenpaaren hinweg übersehen. Langzeit-Sequenzierungstechnologie bietet vielversprechende Möglichkeiten zur zuverlässigen Erkennung neuartiger SVs. BAFME, eine autosomal-dominante neurologische Erkrankung, die durch zitternde Myoklonien und seltene Anfälle gekennzeichnet ist, steht im Zusammenhang mit Wiederholungserweiterungen in der SAMD12 Gen. PacBio SMRT-Sequenzierung, in der Lage, >10-kb DNA zu lesen, hat das Potenzial, die vollständige Abdeckung der SAMD12 Wiederholungs-Expansion. Langzeitlesung Whole-Genome-Sequenzierung Die Verwendung von PacBio kann nützlich sein, um bekannte und neuartige pathogene strukturelle Varianten (SVs) zu erkennen, selbst bei niedriger Abdeckung.
Methoden
- Fünf Familienmitglieder mit BAFME
- Periphere Blutleukozyten
- Genomische DNA-Extraktion
- SMRTbell-Bibliotheksvorbereitung
- SMRT-Sequenzierung
- Sequel-Sequenzierkit 2.0.
- SMRT-Analysemodul
- Southern-Blot-Analyse
- Punktediagrammanalyse
Ergebnisse
In einer vier Generationen umfassenden japanischen Familie, die von BAFME betroffen ist, wurde eine heterozygote strukturelle Variante (SV) an der SAMD12 In der betroffenen Person wurde eine intronische Wiederholungsregion identifiziert, die mit früheren Studien übereinstimmt. Die Größe der Wiederholungsdehnung war zwischen den Individuen nach dem väterlichen Keimbahnübergang ähnlich. Die PacBio SMRT-Sequenzierung konnte die beobachtete Wiederholungsgröße von 4 kb vollständig abdecken. Die Langzeit-Whole-Genome-Sequenzierung (WGS) mit dem Pacbio Sequel-System offenbarte zahlreiche Insertionen und Deletionen, einschließlich sechs SVs, die spezifisch für die betroffene Person in der mit BAFME1 verbundenen Region waren. Eine bemerkenswerte Insertion von 4661 bp wurde zwischen den repetitiven Sequenzen AluSq2 und (TAAAA)n identifiziert. Die Analyse zeigte, dass diese Insertion eine neuartige Sequenz und keine tandemduplizierte Sequenz umfasste, wobei ein hoher Anteil aus Sequenzen mit niedriger Komplexität bestand.
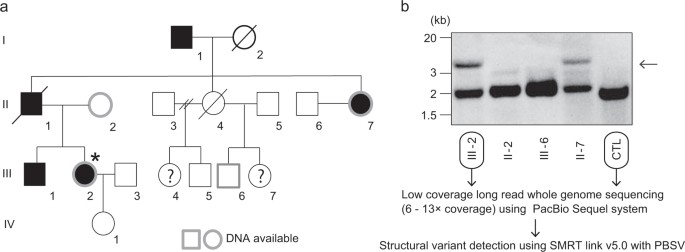 Abb. 1. Stammbaum einer Familie mit pathogener struktureller Variation von SAMD12.
Abb. 1. Stammbaum einer Familie mit pathogener struktureller Variation von SAMD12.
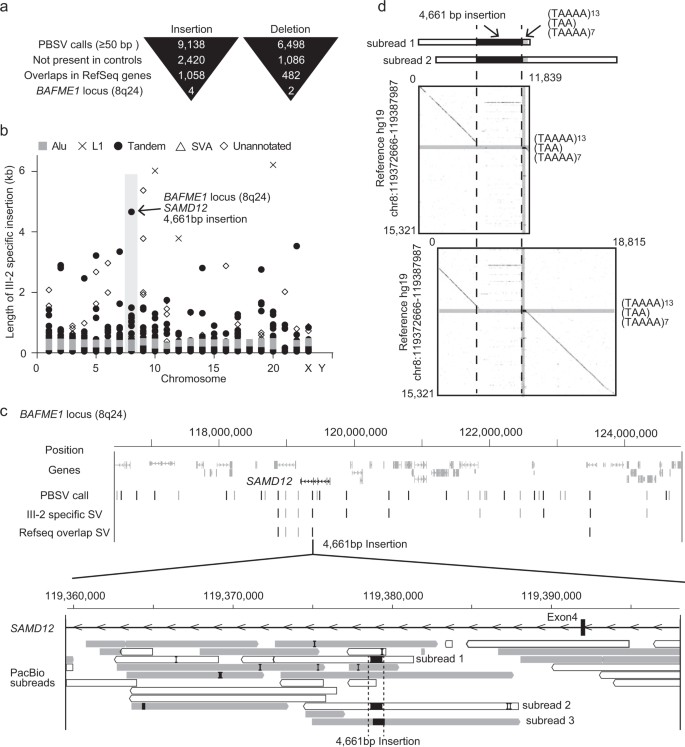 Abb. 2. Bewertung von Langzeit-WGS.
Abb. 2. Bewertung von Langzeit-WGS.
Fazit
Die Langzeit-Sequenzierung liest jetzt DNA über 10 kb und ermöglicht die Erkennung großer struktureller Variationen. Die Autoren wendeten die PacBio SMRT-Sequenzierung auf eine Familie mit gutartiger familiärer Myoklonusepilepsie im Erwachsenenalter an und identifizierten sechs SVs in einer 7,16-Mb BAFME1-Region und bestätigten eine 4,6-kb SAMD12 Wiederholte Einfügungen als Ursache. Langzeit-WGS bietet vielversprechende Möglichkeiten für eine umfassende SV-Analyse und das Aufdecken neuer krankheitsverursachender Varianten bei undiagnostizierten Erkrankungen.
Referenz:
- Mizuguchi T, Toyota T, Adachi H, u. a.Erkennung einer langen Insertion-Variante in SAMD12 durch SMRT-Sequenzierung: Auswirkungen der Langzeit-Whole-Genome-Sequenzierung auf Krankheiten mit Wiederholungserweiterungen. Journal für Humangenetik2019, 64(3):191-7.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Bakterielle Gemeinschaften von Cassiopea in den Florida Keys teilen sich wichtige bakterielle Taxa mit Korallenmikrobiomen.
Journal: bioRxiv
Jahr: 2024
Produktion eines bakteriozinähnlichen Proteins PEG 446 aus Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotika und antimikrobielle Proteine
Jahr: 2024
Entwirrung der Rolle von Pathobionten aus Bacteroides-Arten bei entzündlichen Darmerkrankungen
Journal: bioRxiv
Jahr: 2023
Eine Chromosomenebene-Genomressource zur Untersuchung von Virulenzmechanismen und der Evolution des Kaffeerostpathogens Hemileia vastatrix
Journal: bioRxiv
Jahr: 2022
Streptomyces buecherae sp. nov., ein Actinomycet, der aus mehreren Fledermausarten isoliert wurde
Journal: Antonie van Leeuwenhoek
Jahr: 2020
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


