
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Degradom-Sequenzierung
CD Genomics kann jetzt den Degradom-Sequenzierungsdienst anbieten, um einen umfassenderen Einblick in die Landschaft der pflanzlichen Mikro-RNAs zu ermöglichen. Mit unserem Service können Sie die mRNA-Ziele der Mikro-RNAs auf eine hochsensible und genaue Weise nachweisen.
Die Einführung der Degradom-Sequenzierung
Mikro-RNAs (miRNAs) sind eine Klasse von endogenen nicht-kodierenden RNAs mit einer Länge von 20-24 Nukleotiden (nt), die durch hochpräzise Exzision aus Stem-Loop-Vorläufern produziert werden. Mikro-RNAs sind wichtige Regulatoren der Genexpression auf transkriptioneller und posttranskriptioneller Ebene. Die reife miRNA wird in einen RNA-induzierten Silencing-Komplex (RISC) rekrutiert, um mRNA-Ziele abzubauen und deren Translation zu unterdrücken. miRNAs sind aufgrund von Vergleichen zwischen verschiedenen Arten hochkonserviert. Es gibt auch nicht-konservierte und artspezifische miRNAs in Pflanzen. Nicht-konservierte miRNAs werden oft in niedrigen Mengen exprimiert, und daher werden viele in kleineren Sequenzierungsprojekten nicht identifiziert. Kleine RNA-Sequenzierung Die Technologie hat die Identifizierung von miRNAs mit geringer Häufigkeit ermöglicht.
Modifiziertes 5' RACE (schnelle Amplifikation von cDNA-Enden) wurde häufig zur Bestätigung von Zielen und zur Kartierung von Schnittstellen verwendet. Dennoch ist dieser Ansatz arbeitsintensiv, zeitaufwändig, kostspielig und nur für feingliedrige Untersuchungen anwendbar. Kürzlich hat sich das Degradom-Sequenzieren, auch bekannt als parallele Analyse von RNA-Enden (PARE), als eine leistungsstarke Methode herausgestellt, die beides kombiniert. Hochdurchsatz-Sequenzierung und modifizierte 5' RACE, um nach miRNA und ta-siRNA zu suchentrans-acting siRNA) Ziele in großem Maßstab. Bei dieser Methode wird degradiertes, gekapptes mRNA adapter-ligiert und rücktranskribiert. Die Fragmente werden dann mit Mmel verdaut, gereinigt, 3'-Adapter-ligiert und PCR-amplifiziert. Die Tiefensequenzierung der cDNA und die Degradom-Analyse liefern umfassende Informationen über ungecapte Transkripte, die in Pflanzen degradiert werden.
Vorteile der Degradom-Sequenzierung
- Identifizierung bekannter und neuer miRNAs und ta-siRNAs
- Identifizierung von miRNA-Zielen und regulatorischen Netzwerken
- Statistische Zusammenfassung der mRNA-Abbaustellen
- Untersucht neuartige Biomarker und regulatorische Netzwerke von circRNAs
- Hochdurchsatz und hochauflösend
Degradom-Sequenzierungs-Workflow
Der allgemeine Arbeitsablauf für Degradom-Sequenzierung ist unten skizziert. Um eine Degradom-Sequenzierungsbibliothek zu erstellen, besteht der erste Schritt darin, PolyA-RNA-Proben mit einem RNA-Adapter zu ligieren, der eine 3'Mme I-Stelle enthält, und diese zu transkribieren. Nach der Synthese des zweiten Strangs, der Mme I-Digestion, der Gelreinigung und der PCR-Amplifikation wird der 3'-Adapter für die Tiefensequenzierung ligiert. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und überwacht jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten.

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
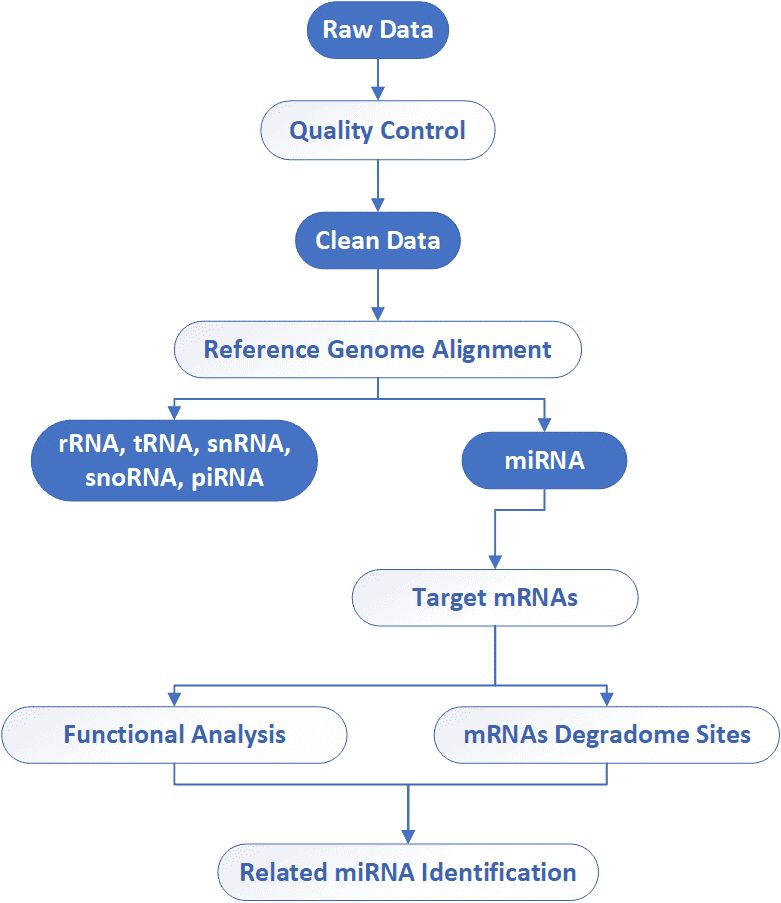
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Degradom-Sequenzierung für Ihr Schreiben (Anpassung)
Unterstützt von unseren erfahrenen Wissenschaftlern und fortschrittlicher Technologie kann CD Genomics Ihnen helfen, kleine RNA-Ziele mit einer Einzelbasenauflösung gleichzeitig zu identifizieren durch die Hochdurchsatz-Sequenzierung durch strenge Qualitätskontrollen und fortschrittliche bioinformatische Analysen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
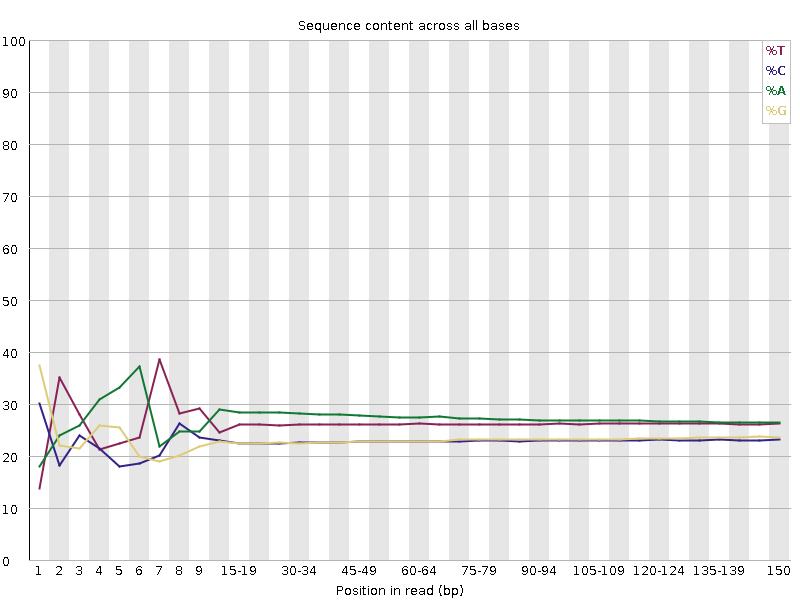
A/T/G/C-Verteilung
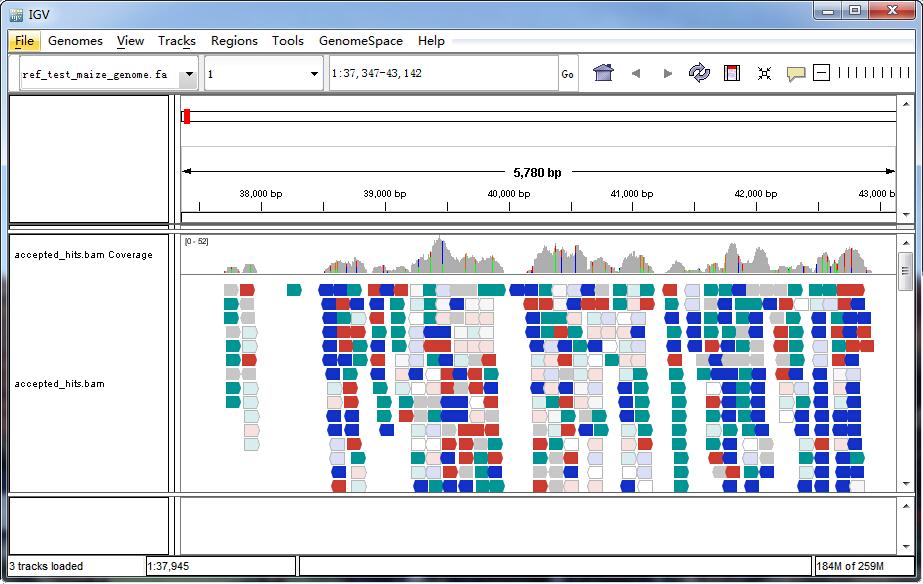
IGV-Browser-Oberfläche
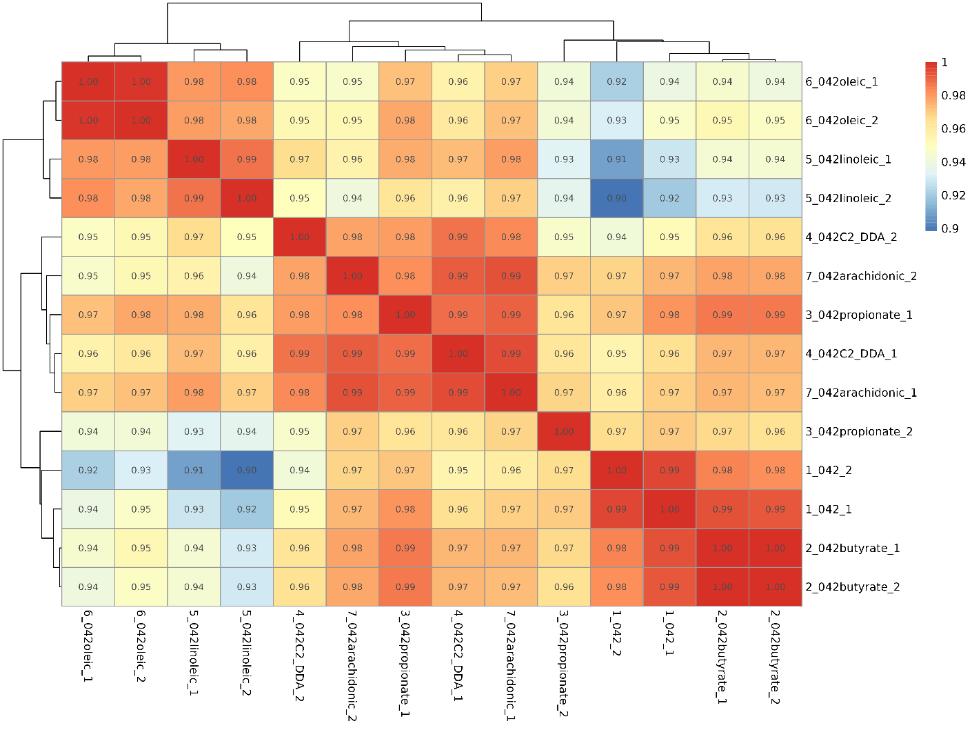
Korrelationsanalyse zwischen Proben
PCA-Score-Diagramm

Venn-Diagramm
Vulkan-Diagramm
Statistik Ergebnisse der GO-Annotierung
KEGG-Klassifikation
Degradome Seq FAQs
1. Was ist das Prinzip der Degradom-Sequenzierung?
Bei Tieren wird angenommen, dass die Proteinrepression durch translationale Hemmung sowie durch mRNA-Abbau erfolgt. Bei Pflanzen degradieren miRNAs ihre mRNA-Ziele durch präzise Spaltung zwischen dem 10. und 11. Nukleotid vom 5'-Ende der miRNA im komplementären Bereich des Zieltranskripts, wodurch ein ausgeprägter Peak des Degradom-Sequenz-Tags an der vorhergesagten Spaltungsstelle im Vergleich zu anderen Regionen des Transkripts entsteht. Basierend auf diesem Prinzip wird die Degradom-Sequenzierung hauptsächlich verwendet, um global Überreste von durch kleine RNA geleiteten Spaltungen zu identifizieren, indem die 5'-Enden von nicht gekappten RNAs in Pflanzen sequenziert werden.
2. Was sind die Vorteile der Degradom-Sequenzierung zur Identifizierung von miRNA-Zielen?
Tabelle 1. Der Vergleich von Degradom-Sequenzierung und traditionellen Methoden zur Vorhersage von miRNA-Zielen.
| Degradom-Sequenzierung | Luciferase-Reporter-Gen-Assay | Argonaute-RNA-Immunopräzipitation (AGO-RIP) | 5' RACE (schnelle Amplifikation der cDNA-Enden) | |
| Durchsatz | Hoch | Niedrig | Niedrig | Niedrig |
| Betrieb | Einfach | Kompliziert | Kompliziert | Einfach |
| Periode | Kurz | Lang | Lang | Lang |
| Genauigkeit | Hoch | Hoch | Relativ hoch | Relativ hoch |
3. Wie unterscheidet sich die Degradom-Sequenzierung von anderen RNA-Sequenzierungsmethoden?
Degradom-Sequenzierung konzentriert sich speziell auf RNA-Abbauprodukte, während andere RNA-Sequenzierungsmethoden wie RNA-Seq Typischerweise wird das gesamte Transkriptom analysiert. Dies macht die Degradom-Sequenzierung besonders geeignet für das Studium von miRNA/siRNA-vermittelten RNA-Spaltungsevents.
Referenz
- Riffo-Campos, Á.L., u. a.Werkzeuge zur sequenzbasierten Vorhersage von miRNA-Zielen: Was soll man wählen? Internationale Zeitschrift für Molekularwissenschaften, 2016, 17(12): 1987.
Degradome-Seq Fallstudien
Kleine RNA- und Degradom-Sequenzierung zeigt wichtige MikroRNA-Funktionen in Astragalus chrysochlorus Reaktion auf Selenstimuli
Journal: Pflanzenbiotechnologie-Journal
Impactfaktor: 6,305
Veröffentlicht: 21. Mai 2015
Zusammenfassung
Astragalus Arten, die als Hyperakkumulatoren von Se bekannt sind, indem sie es in nicht-amino-säurehaltige Verbindungen umwandeln. Aber wir haben keine Ahnung über die mit dem Se-Stoffwechsel verbundene Hyperakkumulation. Die Autoren versuchten zu verstehen, ob miRNAs eine Rolle bei der Se-Akkumulation in Pflanzen spielen. In dieser Studie identifizierten sie 418 bekannte miRNAs und 151 neuartige miRNAs, die durch Se-Exposition induziert wurden in Astragalus chrysochlorusDurch tiefgehende Degradom-Sequenzierung enthüllten die Autoren wichtige miRNA-Funktionen in Ein. Chrysochlorus Reaktion auf Selenstimuli.
Materialien & Methoden
Proben
Kallusgewebe von Ein. Chrysochlorus Samen;
Selenbehandlung.
Sequenzierung
RNA-Isolierung
RNA-Qualitäts- und -Mengenmessungen
Kleine RNA-Sequenzierung
Degradom-Sequenzierung
miRNA-Identifizierung
Zielvorhersage
Funktionsklassifizierung basierend auf GO- und KEGG-Analysen
Ergebnisse
1. miRNA-Identifikation und Expressionsprofile.
Insgesamt wurden 418 bekannte und 151 neuartige miRNAs identifiziert. Die Verteilung der miRNAs zwischen der Kontroll- und der Se-Behandlung ist in Abbildung 1 dargestellt. Die 418 miRNAs gehören zu 380 miRNA-Familien, und 160 miRNA-Familien wurden in beiden Proben (Kontrolle und Se-behandelt) unterschiedlich exprimiert. 30 neuartige miRNAs wurden nach der Se-Behandlung unterschiedlich exprimiert.
 Abbildung 1. Verteilung der miRNAs zwischen Kontrolle und Se-Behandlung: (a) konservierte miRNAs; (b) neuartige miRNAs.
Abbildung 1. Verteilung der miRNAs zwischen Kontrolle und Se-Behandlung: (a) konservierte miRNAs; (b) neuartige miRNAs.
 Abbildung 2. Kleine RNA-Expressionsprofile von Kontroll- und mit Se-behandeltem Kallus von Ein. Chrysochlorus.
Abbildung 2. Kleine RNA-Expressionsprofile von Kontroll- und mit Se-behandeltem Kallus von Ein. Chrysochlorus.
 Abbildung 3. Expressionsprofile von zufällig ausgewählten miRNAs mit unterschiedlicher Häufigkeit in Se-behandelten Ein. Chrysochlorus Insgesamt wurden 1339 vorhergesagte Stellen identifiziert, die von 499 miRNAs geschnitten werden. Die Zielgene wurden annotiert und als Transkriptionsfaktoren und deren Untereinheiten, enzymkodierende Gene, Resistenzproteine, Leucin-reiche Wiederholungen, Leucinzipper, Zinkfingerproteine sowie andere strukturelle und funktionelle Proteine klassifiziert.
Abbildung 3. Expressionsprofile von zufällig ausgewählten miRNAs mit unterschiedlicher Häufigkeit in Se-behandelten Ein. Chrysochlorus Insgesamt wurden 1339 vorhergesagte Stellen identifiziert, die von 499 miRNAs geschnitten werden. Die Zielgene wurden annotiert und als Transkriptionsfaktoren und deren Untereinheiten, enzymkodierende Gene, Resistenzproteine, Leucin-reiche Wiederholungen, Leucinzipper, Zinkfingerproteine sowie andere strukturelle und funktionelle Proteine klassifiziert.
 Abbildung 4. Zielplots (t-Plots) von miRNAs und ihren Zielen. Die roten Pfeile zeigen die häufigsten Spitzen oder Spaltstellen an. (a) miR162-3p zielt auf das Endonuklease Dicer-Homolog-1-ähnliche Protein; (b) miR1513a zielt auf das blau licht-aktivierte Histidin-Kinase; (c) miR2118b zielt auf das Hypoxanthin-Guanin-Phosphoribosyltransferase-ähnliche Protein; (d) miR172c zielt auf das putative ethylen-responsive Transkriptionsfaktor RAP-2-7-ähnliche Protein; (e) miR159b-3p zielt auf ein hypothetisches Protein 11M9.5; (f) miR166 h-3p zielt auf das Homeobox-Leucin-Zipper-Protein ATHB-15-ähnliches Protein.
Abbildung 4. Zielplots (t-Plots) von miRNAs und ihren Zielen. Die roten Pfeile zeigen die häufigsten Spitzen oder Spaltstellen an. (a) miR162-3p zielt auf das Endonuklease Dicer-Homolog-1-ähnliche Protein; (b) miR1513a zielt auf das blau licht-aktivierte Histidin-Kinase; (c) miR2118b zielt auf das Hypoxanthin-Guanin-Phosphoribosyltransferase-ähnliche Protein; (d) miR172c zielt auf das putative ethylen-responsive Transkriptionsfaktor RAP-2-7-ähnliche Protein; (e) miR159b-3p zielt auf ein hypothetisches Protein 11M9.5; (f) miR166 h-3p zielt auf das Homeobox-Leucin-Zipper-Protein ATHB-15-ähnliches Protein.
3. GO- und KEGG-Pfadanalysen
Die Ziele der identifizierten miRNAs wurden einer GO- und KEGG-Analyse unterzogen, um ihre biologischen Rollen zu verstehen. Die Zielgene sind an 47 Arten von Zellkomponenten, 103 Arten von molekularen Funktionen und 144 Arten von biologischen Prozessen beteiligt.
 Abbildung 5. GO-Klassifikationen der miRNA-Ziele in Ein. Chrysochlorus.
Abbildung 5. GO-Klassifikationen der miRNA-Ziele in Ein. Chrysochlorus.
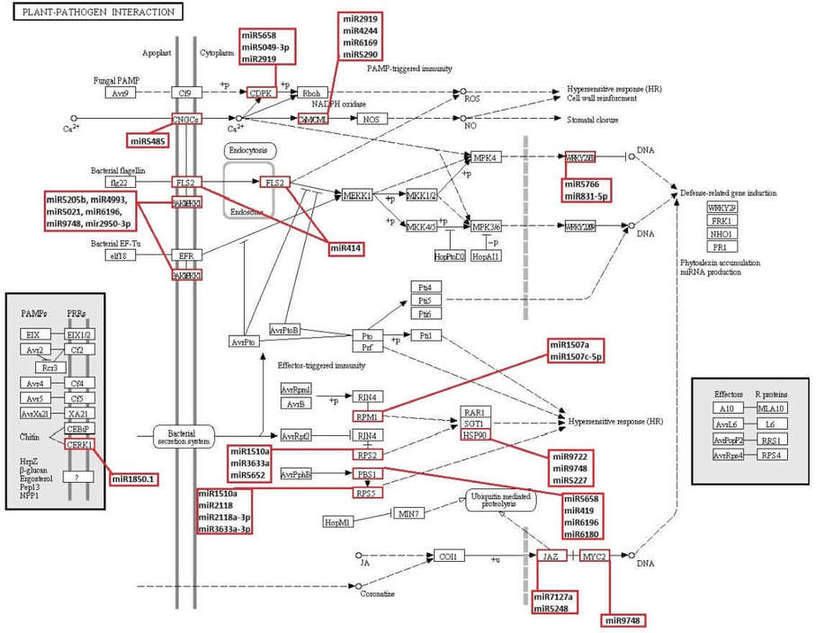 Abbildung 6. KEGG-Pflanzen-Pathogen-Interaktionsweg und neuartige sowie bekannte miRNAs, die in dieser Studie identifiziert wurden und möglicherweise die in diesem Weg beteiligten Gene anvisieren.
Abbildung 6. KEGG-Pflanzen-Pathogen-Interaktionsweg und neuartige sowie bekannte miRNAs, die in dieser Studie identifiziert wurden und möglicherweise die in diesem Weg beteiligten Gene anvisieren.
Referenz
- Cakir O, Candar-Cakir B, Zhang B. Die Sequenzierung von kleinen RNAs und Degradomen zeigt eine wichtige Funktion von Mikro-RNAs in Astragalus chrysochlorus Reaktion auf Selenstimuli. Pflanzenbiotechnologie-Journal, 2016, 14(2): 543-556.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege, um die Aktivität von Gliom-Stammzellen aufrechtzuerhalten.
Journal: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 bei Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikationen vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzell-Eigenschaft und die Bildung von Blinddarmtumoren zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neonatalen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


