
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben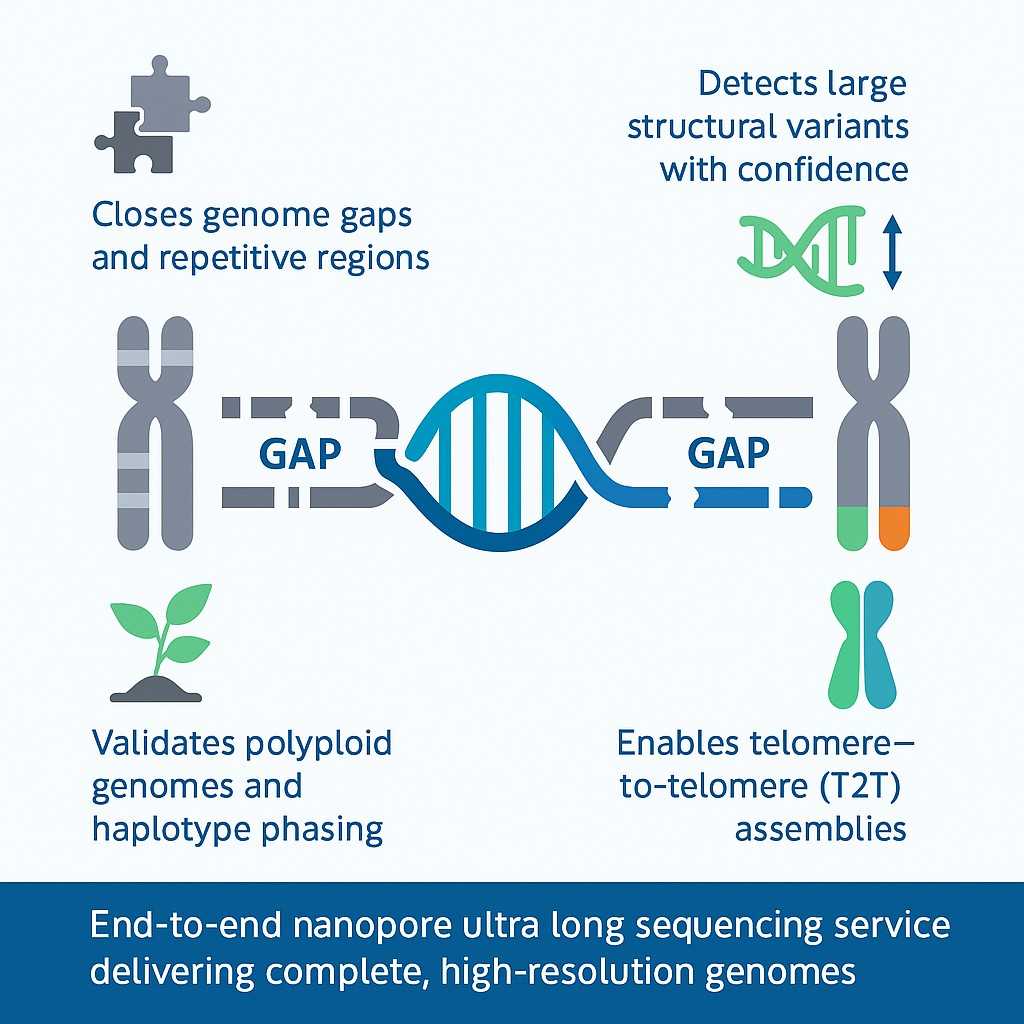
Warum ultralange Nanoporen-Sequenzierung wichtig ist
Konventionelles Sequencing hinterlässt oft ungelöste Lücken, insbesondere in Genomen mit umfangreichen Wiederholungen, Polyploidie oder hoher struktureller Komplexität. Diese Lücken beeinträchtigen die Genauigkeit von Genomassemblierungen und verringern das Vertrauen in nachgelagerte Analysen.
Nanopore Ultra-Langsequenzierung adressiert diese Einschränkungen, indem sie DNA-Reads erzeugt, die weit länger sind als bei Standardansätzen. Im Gegensatz zur Kurz- oder Mittel-Längen-Sequenzierung, die Schwierigkeiten mit stark repetitiven Regionen hat, können ultra-lange Reads Zentromere, Telomere und strukturelle Varianten in einem einzigen Abschnitt abdecken.
Diese Fähigkeit hat die Genomforschung revolutioniert. Pflanzen- und Tierstudien erreichen jetzt routinemäßig Telomer-zu-Telomer (T2T) Assemblierungen, was es Forschern ermöglicht, die genetische Architektur mit beispielloser Auflösung zu erforschen. Für angewandte Forschung in der Pflanzenzüchtung, biomedizinischen Entdeckung und mikrobiellen Evolution bietet die Fähigkeit, vollständige Genome zu entschlüsseln, einen direkten Wettbewerbsvorteil.
 Die Ganzgenomsequenzierung deckt Virulenzfaktoren, mobile genetische Elemente und potenzielle Umweltübertragungen von Bakterienstämmen in Rinderhaltungsumgebungen auf. (Rivu, Supantha, et al., 2024)
Die Ganzgenomsequenzierung deckt Virulenzfaktoren, mobile genetische Elemente und potenzielle Umweltübertragungen von Bakterienstämmen in Rinderhaltungsumgebungen auf. (Rivu, Supantha, et al., 2024)
Technische Parameter
| Parameter | Spezifikation |
|---|---|
| Leseumfang | N50 >50–100 kb; maximale Reads >4 Mb |
| Eingabebedarf | ≥6 Millionen Zellen (PBMCs, kultivierte Zellen oder gefrorenes Gewebe) |
| Chemie | Nanopore Ultra-Long Sequenzierungs-Kit (SQK-ULK114, Kit 14, R10.4.1 Nanopore) |
| Plattform | PromethION / GridION |
| Durchsatz | Bis zu 90–100 Gb pro PromethION-Flusszelle |
| Genauigkeit | Rohlese Q20+ (Kit 14 Chemie) |
| Vorbereitungszeit | ~200 Minuten plus Übernachtelution |
| QC-Methoden | Qubit, Nanodrop, gepulste Feldgel-Elektrophorese (PFGE) |
| Lagerung & Logistik | Kits werden bei 2–8 °C versendet; langfristige Lagerung bei –20 °C |
Vorteil: CD Genomics Service-Highlights
CD Genomics liefert ein End-to-End-Nanoporen-Ultra-Long-Sequenzierungsdienst das fortschrittliche Laborprotokolle mit bewährten Sequenzierungsplattformen kombiniert. Unser Ansatz ist darauf ausgelegt, die Leseweite, Stabilität und Genauigkeit zu maximieren, sodass Forscher Lücken schließen und hochkontinuierliche Assemblierungen erzeugen können.
Wesentliche Servicevorteile
- Lückenlose Assemblierungen: Ultra-lange DNA-Sequenzierung mit Nanoporen überbrückt sich wiederholende und GC-reiche Regionen und beseitigt Assemblierungslücken.
- Entdeckung struktureller Varianten: Lange Reads erkennen große Insertionen, Deletionen, Inversionen und Wiederholungserweiterungen mit hoher Zuverlässigkeit.
- Polyploide Genomauflösung: Optimierte Strategien validieren die Haplotyp-Phasierung in komplexen Arten.
- Telomer-zu-Telomer-Analyse: Nanopore-Langread-Sequenzierung unterstützt vollständige T2T-Assemblierungen in Pflanzen und Tieren.
- Optimierte Chemie: Die Verwendung des Nanopore Ultra-Long Sequencing Kits (SQK-ULK114) mit Kit 14 gewährleistet eine hohe Ausbeute und eine Rohlesequalität von Q20+.
- Flexible Anwendungen: Kompatibel mit genomischer DNA und integriert mit nachgelagerten Nanoporen-RNA-Sequenzierungslösungen.
- Eigene SOPs zur Extraktion von ultrahochmolekularem DNA aus Pflanzen-, Tier- und Mikrobiologischen Proben.
- Workflow-Verbesserungen bei der DNA-Reparatur und der Bibliothekskonstruktion, die eine Fragmentlänge von über 100 kb erhalten.
- Zugang zu den PromethION- und GridION-Plattformen mit der neuesten Kit 14-Chemie für Q20+-Genauigkeit.
Bewiesene Fallbeweise
| Projekt | Strategie / Datenausgabe | Schlüsselergebnisse |
|---|---|---|
| Weizen-Genomassemblierung | HiFi + ONT ultra lange Reads | Contig N50 verbesserte sich von 341 kb auf 2,15 Mb; nahezu lückenreferenz erreicht |
| Validierung des Zuckerrüben-Genoms | Ultralange Nanoporenlesungen zur Haplotype-Verifizierung | Wechsel-Fehlerquote von bis zu 0,05/Mb; >90% Mapping-Genauigkeit |
| Chili-Pfeffer T2T-Genom | Vier PromethION-Flow-Zellen; N50-Leselänge bis zu 107 kb | Durchschnittliches N50 = 91,5 kb; längste Lesung 2,98 Mb; vollständige T2T-Zusammenstellung |
| Sorghum-Genomassemblierung | ONT Ultra-Long-Sequenzierung nur (kein Illumina/PacBio) | Abgeschlossene Telomer-zu-Telomer-Assemblierung; validierte Zentromere und Telomere |
Hauptanwendungen
De-novo-Genomassemblierung
Ultra-lange Reads überbrücken repetitive Elemente und GC-reiche Regionen und schließen Lücken, die mit Short-Read- oder Standard-Lang-Read-Sequenzierung ungelöst bleiben.
Erkennung struktureller Variationen
Nanopore-Langlesesequenzierung identifiziert große Insertionen, Deletionen, Inversionen und Wiederholungserweiterungen, die die Genomstabilität und den Phänotyp beeinflussen.
Polyploide Genomanalyse
Ultralange DNA-Sequenzierung mit Nanoporen verbessert die Haplotyp-Phasierung und validiert Assemblierungen in polyploiden Pflanzen und hybriden Arten.
Telomer-zu-Telomer (T2T) Assemblierung
Vollständige Chromosomenebene-Assemblierungen werden erreicht, indem Telomere, Zentromere und rDNA-Regionen mit Einzelreads überspannt werden.
Transkriptom und RNA-Sequenzierung Integration
Kombiniert mit Nanopore-Direkt-RNA-Sequenzierung und Nanopore-Voll-Längen-Transkript-SequenzierungForschern ist es möglich, die Genomstruktur mit der transkriptionalen Aktivität zu verknüpfen.
Workflow: End-to-End-Service
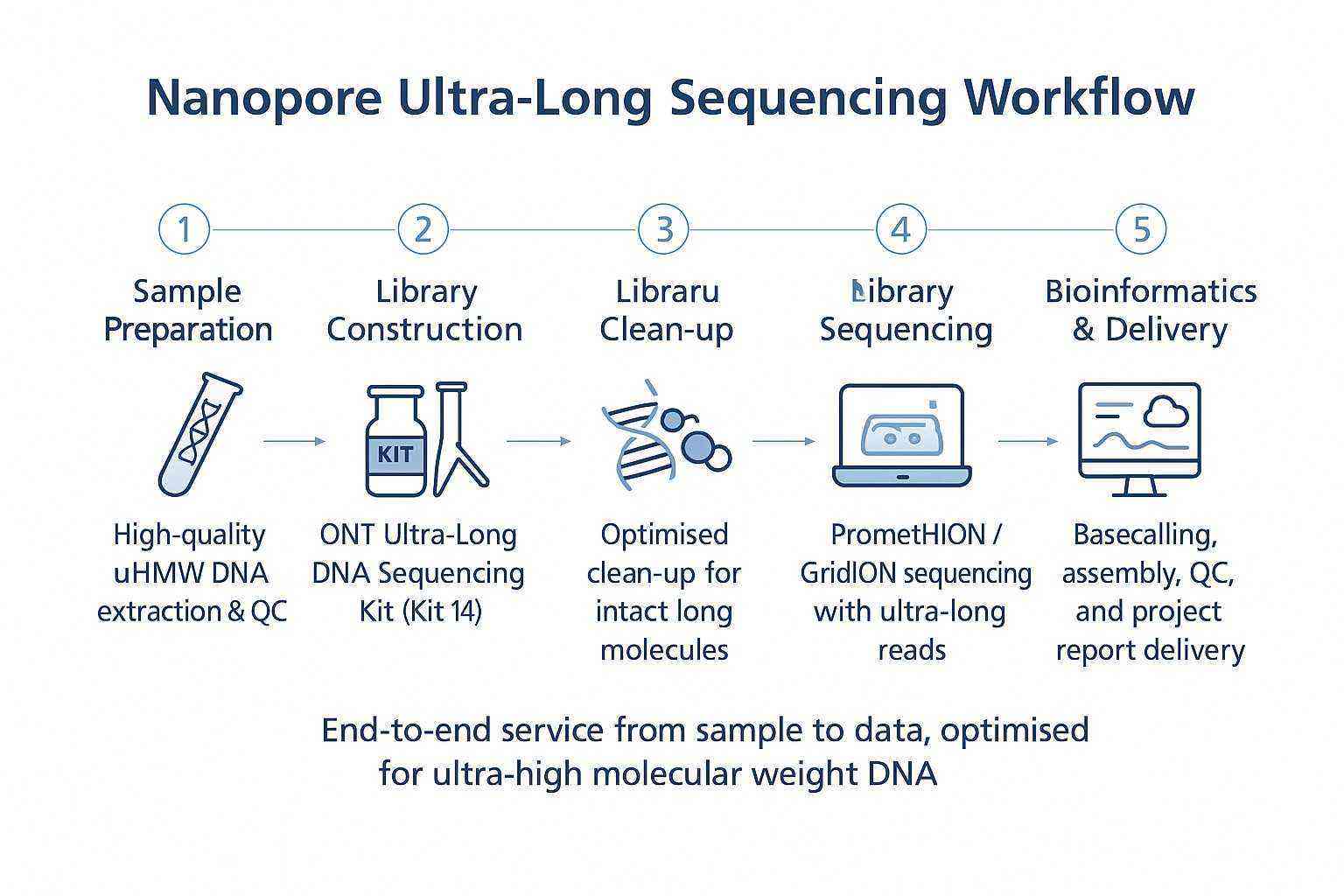
CD Genomics bietet einen vollständigen Workflow für Nanopore-Ultra-Long-Sequenzierungvon der Probenvorbereitung bis zur endgültigen Datenlieferung. Jede Phase ist optimiert, um ultra-hochmolekulare DNA zu erhalten und die Leseweite zu maximieren.
1. Probenvorbereitung
- Unterstützung für Pflanzen-, Tier- und Mikrobiologiemuster
- Spezialisierte Protokolle zur Minimierung der Degradation und zur Entfernung von Polysacchariden oder sekundären Metaboliten
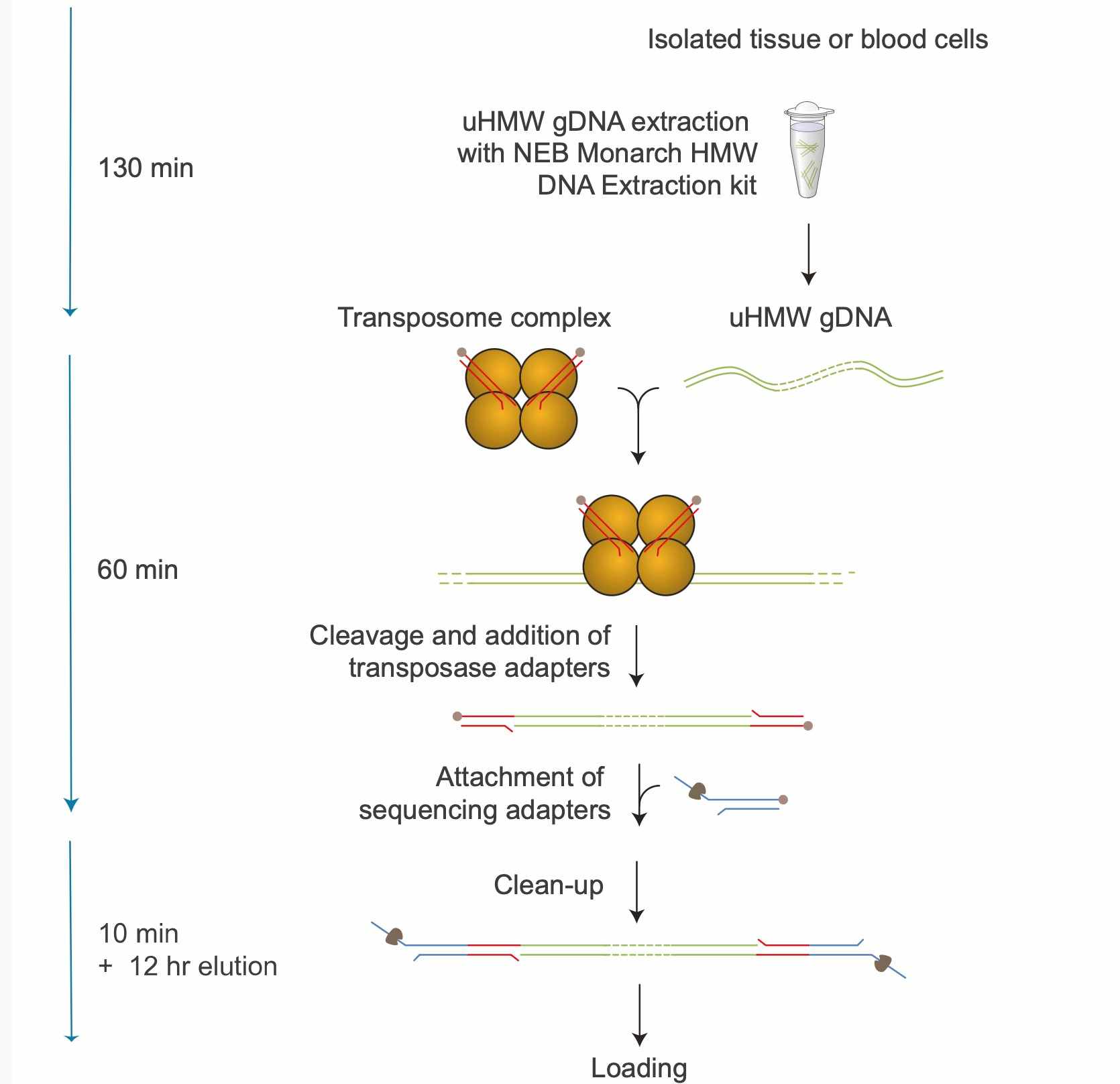
2. DNA-Extraktion und Qualitätskontrolle
- Extraktion von ultrahochmolekularem Gewicht (uHMW) DNA
- Qualitätskontrollen mit Qubit, Nanodrop und Pulsfeld-Gelelektrophorese
3. Bibliotheksvorbereitung
- Nanopore Ultra-Long Sequenzierungs-Kit (SQK-ULK114) mit Kit 14 Chemie
- Transposase-basierte Fragmentierung und schnelle Adapterligierung
- Übernachtige Elution zur Erhaltung langer DNA-Moleküle
4. Sequenzierung
- Durchgeführt auf den PromethION- oder GridION-Plattformen
- Erreichen Sie Lese-Längen >100 kb N50, mit maximalen Lese-Längen über 4 Mb.
5. Bioinformatische Analyse
- Echtzeit-Basiscalling und Qualitätsfilterung
- Montage, Polieren und Variantenkennung
- Optionale Telomer-zu-Telomer (T2T) Assemblierungsunterstützung
6. Berichterstattung und Lieferung
- FASTQ- und BAM-Dateien mit QC-Metriken
- Angepasste Analyseberichte für Genomassemblierung oder Studien zu strukturellen Varianten
Bioinformatische Analyse
Grundlegende Analyse
| Analysephase | Beschreibung |
|---|---|
| Basisabgleich | Konvertiert rohe elektrische Signale in DNA/RNA-Sequenzen mithilfe von Modellen wie Dorado für verbesserte Genauigkeit. |
| Qualitätskontrolle (QC) | Beinhaltet die Verteilung der Lese-Längen, Abdeckungsmetriken und Qualitätswerte. |
| De novo-Assembly | Erstellt hochkontinuierliche genomische Assemblierungen mit Werkzeugen, die für lange Reads optimiert sind (z. B. Flye, Canu). |
| Polieren und Fehlerkorrektur | Verfeinert die Fehlerprofile der Assemblierung mithilfe von Long-Read-Selbstkorrektur oder hybriden Polier-Workflows. |
Fortgeschrittene Analyse
| Analysephase | Beschreibung |
|---|---|
| Strukturelle Variantenbestimmung | Erkennt große Indels, Duplikationen und Inversionen mit Tools wie Sniffles oder CuteSV. |
| Variant-Phasierung & SV-Annotation | Phasenvarianten über lange Contigs hinweg und annotiert SVs für die biologische Interpretation. |
| Telomere-zu-Telomere (T2T) Unterstützung | Vollständigt Chromosomenebene-Assemblierungen, indem Lücken an Wiederholungen, Zentromeren und Telomeren geschlossen werden. |
| Epigenetische und Basenmodifikationserkennung | Erkennt DNA/RNA-Modifikationen (z. B. 5mC, m6A) mithilfe von Remora oder Megalodon während der Basenbestimmung. |
| Metagenomische / Taxonomische Klassifikation | Klassifiziert Lesevorgänge in gemischten Proben mithilfe von Workflows wie den EPI2ME-Metapipelines. |
Liefergegenstände
Kunden erhalten:
- Rohsequenzierungsdaten (FASTQ-Format)
- Qualitätskontrollbericht (Längenverteilung, N50, Ertrag, Genauigkeit)
- Optionale Ergebnisse der Genomassemblierung und Variantenanalyse
- Projektzusammenfassungsbericht
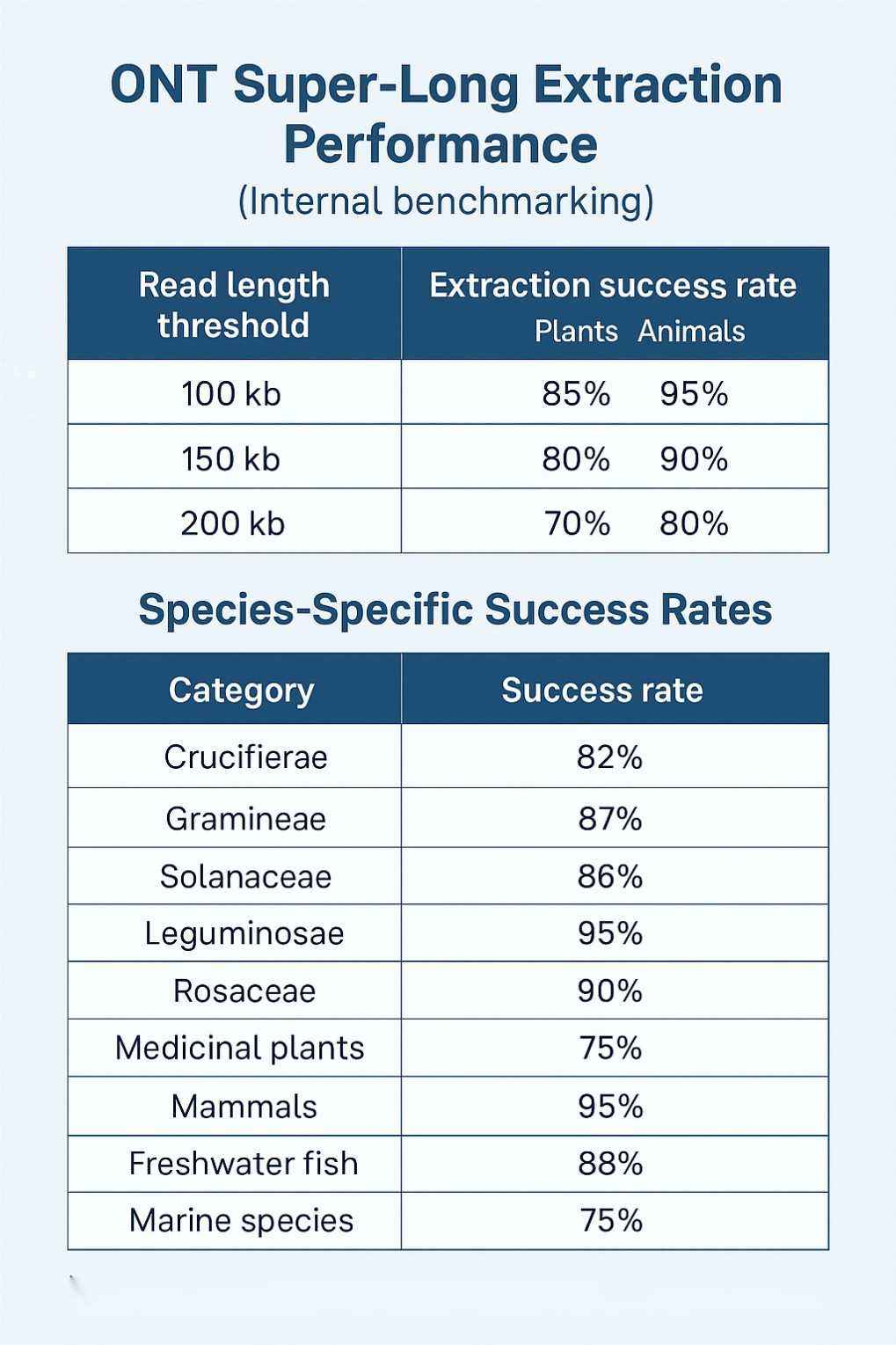
Musteranforderungen für Nanopore Ultra-lange Sequenzierung
Um optimale Sequenzierungsergebnisse zu gewährleisten, benötigen wir die folgenden Probenbedingungen:
| Probenart | Gewebetyp | Anforderung (pro Zelle) | Bemerkungen |
| Tier | Säugetierblut | ≥5 mL |
|
| Kernhaltige Erythrozyten im Blut (Fische, Reptilien, Amphibien, Vögel/Geflügel) | ≥100 μL | ||
| Zellen | ≥6×10⁷ |
|
|
| Eingeweide | ≥0,5 g | Am wenigsten effektiver Proben-Typ, schwer zu erreichen N50 >100 Kb | |
| Muskel | ≥3 g | ||
| Pflanze | Junge zarte Blätter | ≥3 g |
2. Mit sterilem Wasser abspülen 3. Trocknen und Schnellgefrieren |
Unser Team bietet Richtlinien zur Probenvorbereitung, um Ihnen zu helfen, die besten Ergebnisse zu erzielen.
Demonstrationsergebnisse
Längenverteilung der Reads
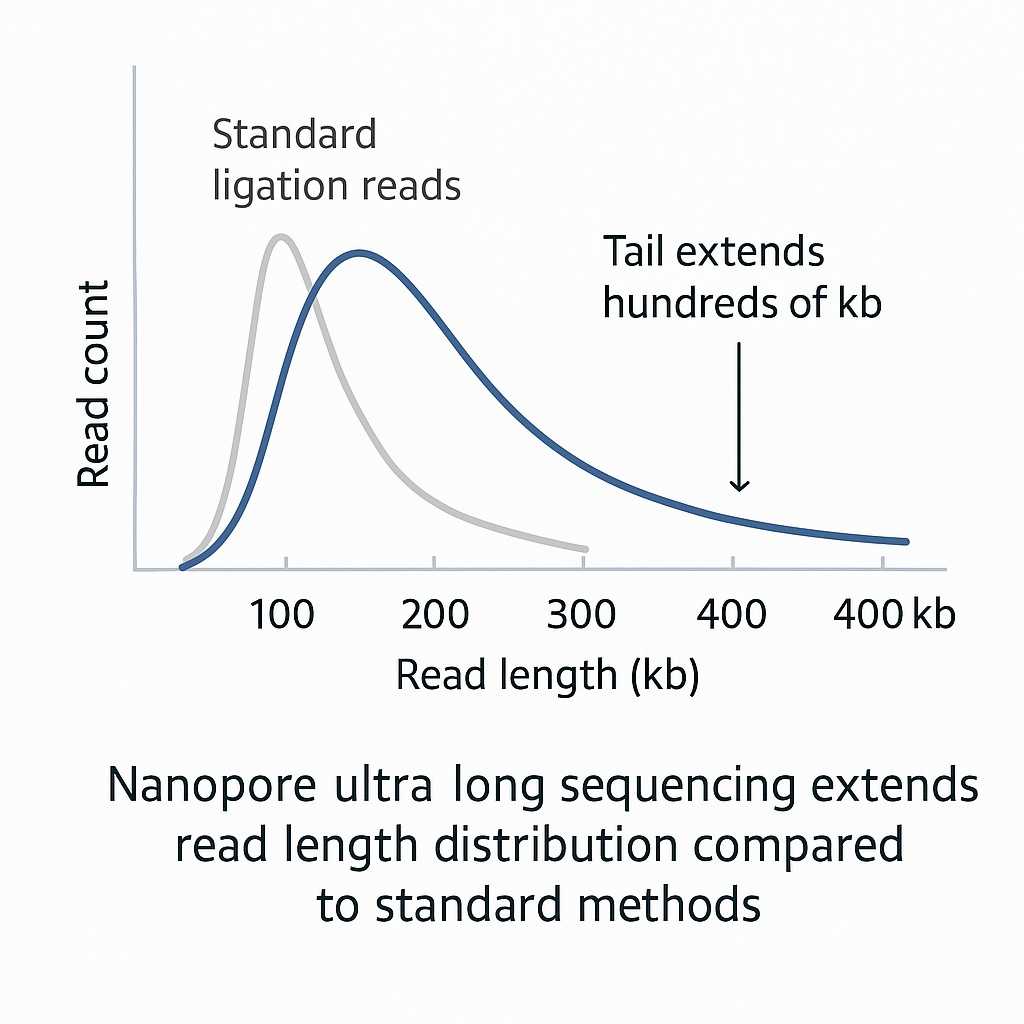
Das beigefügte Diagramm (von Oxford Nanopore Technologies) veranschaulicht, wie ultra-lange Sequenzierung vergrößert dramatisch die Leseweite im Vergleich zu standardmäßigen Ligationstechniken – und erzeugt einen glatten Schwanz, der mehrere hundert Kilobasen erreicht.
Einfluss des Weizengenoms
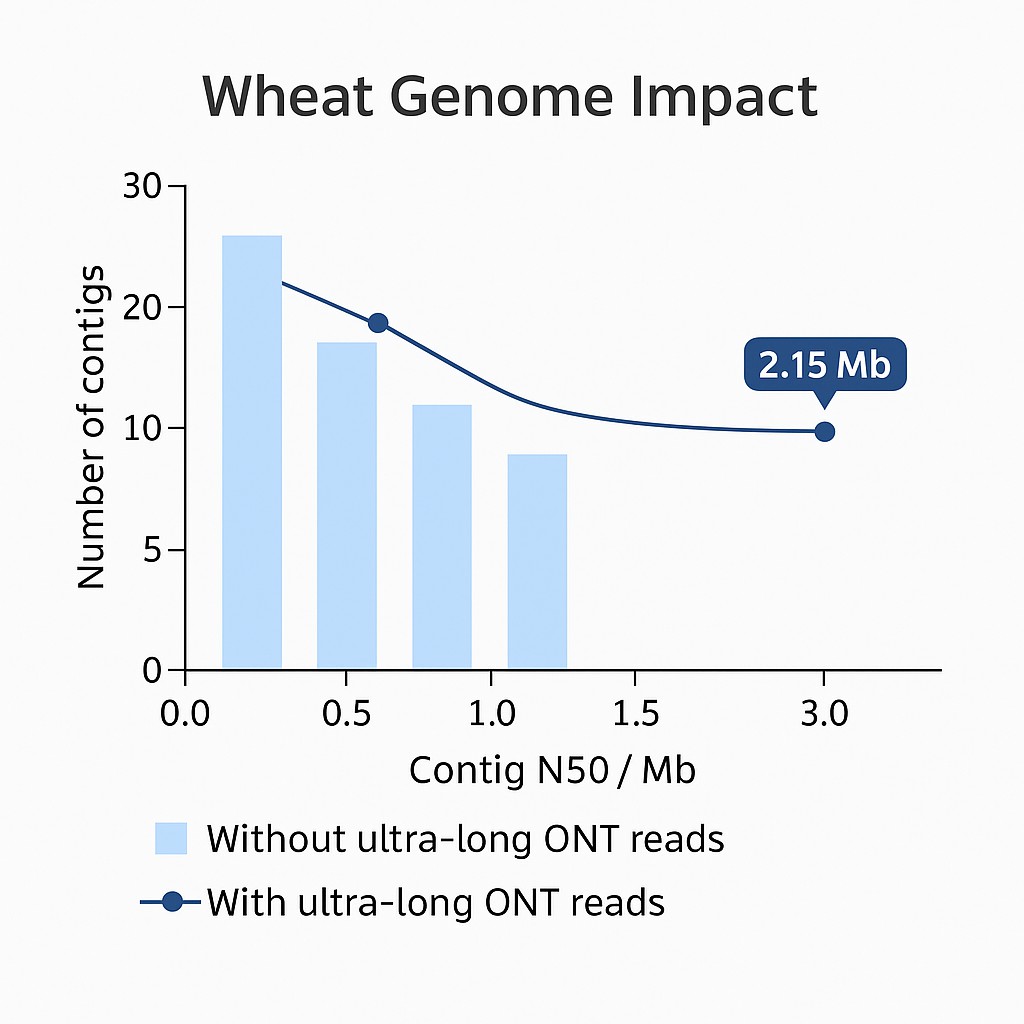
Die Einbeziehung von ultra-langen ONT-Reads erhöhte die Contig N50 von 341 KB auf 2,15 MB, was zu einer nahezu lückenlosen Assemblierung führt, die ideal für nachgelagerte genomische Forschung und Zuchtprogramme ist.
Pflanze T2T-Fortschritt
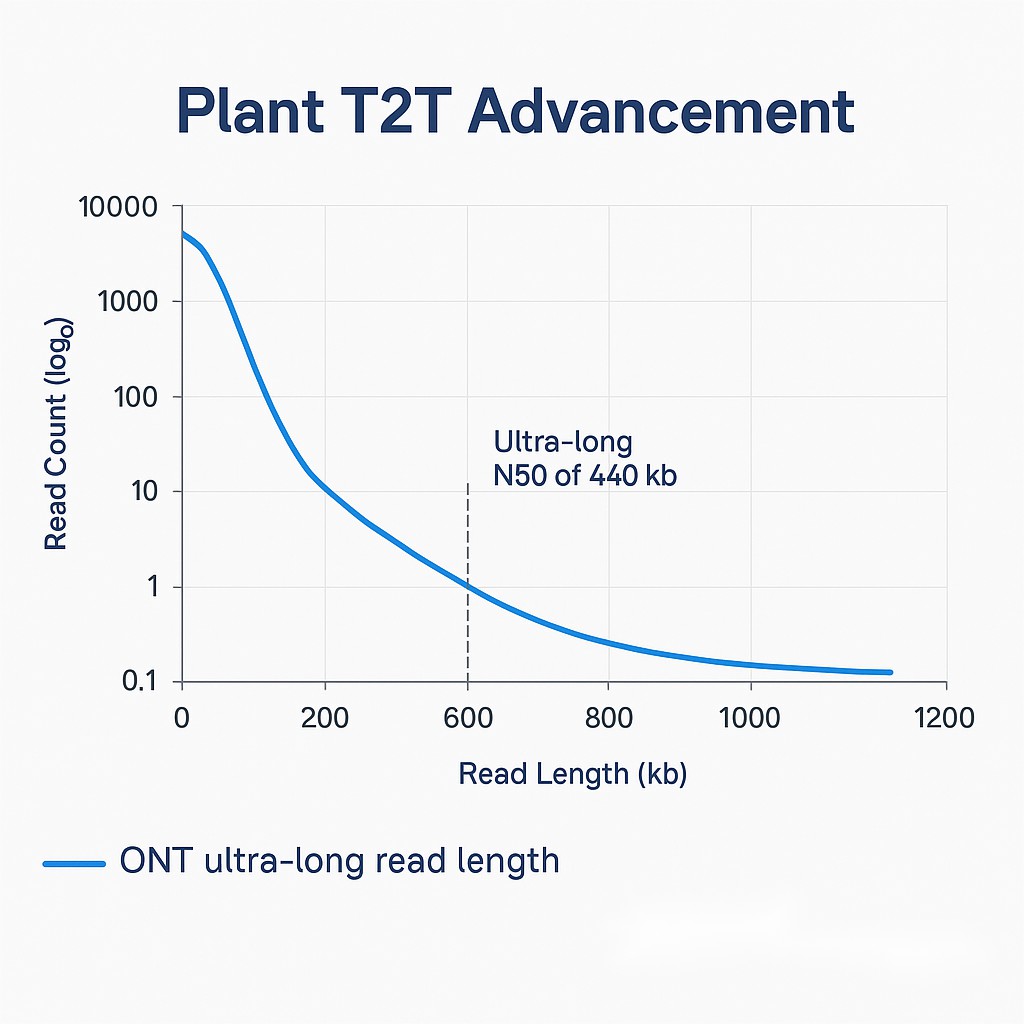
In aktuellen Pflanzen-Genomprojekten lieferten optimierte Extraktions- und Bibliotheksprotokolle ultra-lange Reads mit einem N50 bis zu 440 kb, erheblich automatisiert verbessern Telomer-zu-Telomer (T2T) Versammlung.
Erfolg bei der Sorghum-Assemblierung
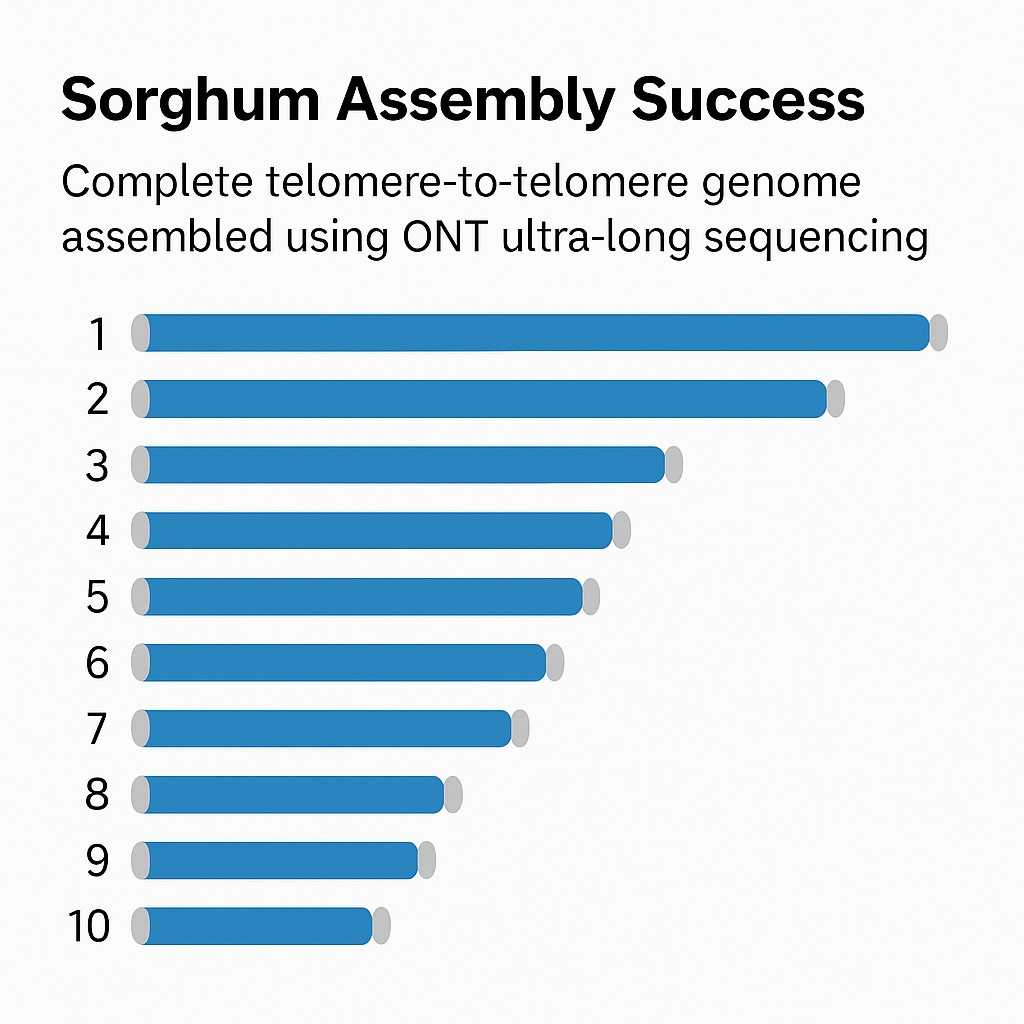
Ein vollständiges T2T Sorghum-Genom wurde ausschließlich mit ONT-Ultra-Langsequenzierung erstellt, was die Fähigkeit der Methode zeigt, vollständige Chromosomen zu entschlüsseln, ohne auf ergänzende Technologien angewiesen zu sein.
Nanopore Ultra-Long Seq FAQs
F: Welche Lese-Länge kann ich von der Nanopore-Ultra-Long-Sequenzierung erwarten?
Sie können N50-Leseweiten erwarten, die typischerweise von 50 bis über 100 kb reichen, und unter optimalen Bedingungen können einzelne Reads 4 Mb überschreiten – ultralange Reads ermöglichen die Auflösung komplexer genomischer Regionen wie Wiederholungen und Zentromere.
F: Warum ist die ultra-lange Leseweite für meine Genomassemblierung wichtig?
Ultra-lange Reads überbrücken hochgradig sich wiederholende oder strukturell komplexe Regionen, was lückenlose oder nahezu lückenlose Assemblierungen wie T2T-Genome ermöglicht und die Erkennung von strukturellen Varianten verbessert, die kürzere Reads nicht auflösen können.
Q: Welchen Einfluss haben lange Reads auf die Erkennung struktureller Varianten?
Lange Nanoporen-Lesungen verbessern die Entdeckung großer Einsätze, Löschungen, Inversionen und Wiederholungserweiterungen, indem sie diese direkt überbrücken, was die Variantenbestimmung vereinfacht und die Mehrdeutigkeit verringert.
Q: Kann die Nanoporen-Sequenzierung sowohl DNA- als auch RNA-Proben verarbeiten?
Ja, die Nanoporen-Sequenzierung unterstützt die direkte Sequenzierung sowohl von DNA- als auch von RNA-Molekülen, ohne dass eine Amplifikation oder Markierung erforderlich ist. Dies ermöglicht es, Transkripte und Basenmodifikationen zusammen mit der Genomstruktur zu untersuchen.
Q: Welche Faktoren beeinflussen die Probenqualität für ultra-lange Reads?
Die DNA-Purität und die Fragmentlänge sind entscheidend. Eine hochwertige Extraktion mit minimaler Degradation ist unerlässlich, und mehrere Extraktionsversuche können erforderlich sein, um DNA mit ultra-hoher Molekulargewicht zu erhalten, die für ultra-lange Lesevorgänge geeignet ist.
Q: Ist die Nanopore-Technologie für den Einsatz im Feld oder für tragbare Anwendungen geeignet?
Ja, weil Nanoporen-Sequenzer native DNA oder RNA in Echtzeit in skalierbaren Formaten verarbeiten können – von tragbaren MinION-Geräten bis hin zu Hochdurchsatzplattformen – unterstützt diese Flexibilität Anwendungen im Labor, im Feld und aus der Ferne.
Nanopore Ultra-Long-Sequenzierungs-Fallstudien
1. Hintergrund
Das menschliche Genom hat eine Größe von etwa 3,1 Gb und enthält umfangreiche repetitive Regionen, segmentale Duplikationen und Heterozygotie, was die Assemblierung mit Kurzlese-Sequenzierung herausfordernd macht. Konventionelle Technologien können Zentromere, Telomere und strukturelle Varianten nicht auflösen, was zu anhaltenden Lücken in Referenzgenomen führt. Diese Fallstudie untersucht, wie Nanopore-Ultra-Long-Sequenzierung kann diese Barrieren überwinden.
2. Methoden
Forscher sequenzierten die GM12878 menschliche Zelllinie (Utah/CEPH-Stammbaum) auf dem Oxford Nanopore MinION Plattform mit R9.4 1D-Chemie. DNA-Vorbereitungsprotokolle wurden entwickelt, um das Scheren zu minimieren und ultra-hochmolekulare Fragmente zu erhalten.
- Generierte Daten: 91,2 Gb Sequenz (~30× Abdeckung) von 39 Flusszellen
- Ultra-lange Reads: N50 >100 kb; maximale Lese-Länge 882 kb
- Montagewerkzeug: Canu-Assembler mit Illumina-Short-Read-Politur zur Verbesserung der Genauigkeit.
3. Ergebnisse
- Nanopore-Only-Assembly NG50 = ~3 Mb
- Hinzufügen von 5× ultra-langer Abdeckung verdoppelt NG50 auf 6,4 Mb
- Haupt-Histokompatibilitätskomplex (MHC) (4 Mb) in einem einzigen Contig aufgelöst
- 12 große Lücken (>50 kb) im GRCh38 wurden geschlossen.
- Endgenauigkeit nach der Nachbearbeitung = 99,88 %
- Ultra-lange Reads ermöglichten die Haplotype-Phasierung über das gesamte MHC-Lokus.
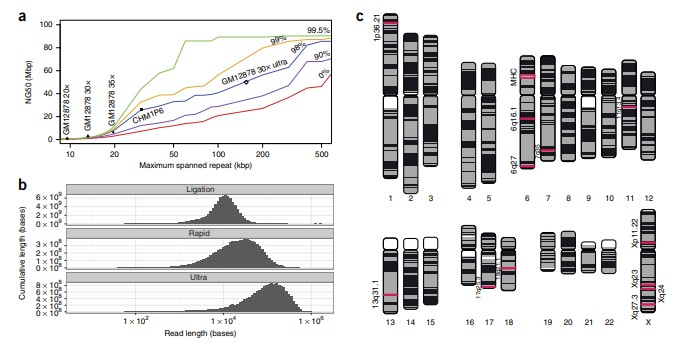 Chromosomenplot, der zeigt, wie die Nanopore-Ultra-Langsequenzierung 12 Lücken im GRCh38 geschlossen hat, einschließlich des 16 Mb MHC-Lokus. Kontinuierliche Farbblöcke repräsentieren zusammenhängende Assemblierungen, während weiße Lücken ungelöste Bereiche anzeigen.
Chromosomenplot, der zeigt, wie die Nanopore-Ultra-Langsequenzierung 12 Lücken im GRCh38 geschlossen hat, einschließlich des 16 Mb MHC-Lokus. Kontinuierliche Farbblöcke repräsentieren zusammenhängende Assemblierungen, während weiße Lücken ungelöste Bereiche anzeigen.
4. Schlussfolgerungen
Diese Studie zeigt, dass Nanopore-Ultra-Long-Read-Sequenzierung ermöglicht hochkontinuierliche menschliche Genomassemblierungen. Die Technologie löste komplexe Loci, schloss Referenzlücken und lieferte Haplotyp-Phasierung auf Chromosomenebene. Diese Ergebnisse heben ihr Potenzial zur Erzeugung nahezu vollständiger Telomer-zu-Telomer (T2T) Assemblierungen hervor und fördern sowohl die grundlegende Genomik als auch translationalen Anwendungen.
Referenzen:
- Lu D, Liu C, Ji W, Xia R, Li S, Liu Y, Liu N, Liu Y, Deng XW, Li B. Nanopore-Ultra-Long-Sequenzierung und adaptive Probenahme fördern die vollständige Telomer-zu-Telomer-Genomassemblierung von Pflanzen.. Mol-Anlage2024 Nov 4;17(11):1773-1786. doi: 10.1016/j.molp.2024.10.008. Epub 2024 Okt 16. PMID: 39420560.
- Prall, T.M., Neumann, E.K., Karl, J.A. et al. Konsistente ultra-lange DNA-Sequenzierung mit automatisierter langsamer Pipettierung. BMC Genomik 22, 182 (2021).
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O'Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M. Nanoporen-Sequenzierung und -Assemblierung eines menschlichen Genoms mit ultralangen Reads. Nat Biotechnol. 2018 Apr;36(4):338-345. doi: 10.1038/nbt.4060. Epub 2018 Jan 29. PMID: 29431738; PMCID: PMC5889714.


