
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
SNP Feinkartierung
CD Genomics bietet einen SNP-Fine-Mapping-Service für eine große Anzahl von SNPs und ein hohes Probenvolumen an, um Ihnen zu helfen, die SNP-Loci von Interesse basierend auf einer Teilmenge der detektierten SNPs zu validieren und zu bestätigen.
Einzelne Nukleotid-Polymorphismen (SNPs) stellen die häufigste Art genetischer Variation zwischen Individuen dar. Feinkartierung, ein fortgeschrittener analytischer Ansatz, ist entscheidend, um die spezifischen SNPs zu identifizieren, die ursächlich Merkmale oder Krankheiten beeinflussen, die durch genomweite Assoziationsstudien (GWAS).

Wofür wird SNP-Kartierung verwendet?
- Identifizierung kausaler Varianten durch SNP-Kartierung
SNP-Kartierung ist eine grundlegende Technik, um kausale Varianten zu erkennen, die für genetische Assoziationen verantwortlich sind, die in GWAS beobachtet werden. Durch die robuste Unterscheidung zwischen bloßen statistischen Assoziationen und tatsächlich kausalen SNPs können Forscher die genetischen Grundlagen komplexer Merkmale und Krankheiten präziser aufklären. Zum Beispiel kann ein GWAS zunächst eine breite genomische Region kennzeichnen, die mit einem bestimmten Krankheitsphänotyp assoziiert ist, während die Feinkartierung diese Auflösung verfeinert, um spezifische SNPs zu identifizieren, die direkt für die beobachtete genetische Assoziation verantwortlich sind. Diese Präzision ist entscheidend für das Vorantreiben unseres Verständnisses der genetischen Architektur von Krankheiten.
- Verbesserung der Interpretation genetischer Assoziationen
Der Prozess des Fine Mappings verbessert die interpretative Kraft genetischer Assoziationen erheblich, indem er systematisch die Liste potenzieller ursächlicher SNPs innerhalb eines bestimmten genomischen Interessengebiets eingrenzt. Dieser gezielte Ansatz ermöglicht präzisere funktionale Studien, die es Forschern erleichtern, die biologischen Mechanismen zu entschlüsseln, durch die genetische Varianten ihren Einfluss auf Phänotypen ausüben. Folglich stellt das Fine Mapping einen entscheidenden Schritt dar, um GWAS-Ergebnisse in umsetzbare biologische Erkenntnisse zu übersetzen.
- Leitende Funktionelle Genomik
Über seine Rolle bei der Verfeinerung genetischer Assoziationen hinaus ist die Feinkartierung entscheidend für den Fortschritt der funktionellen Genomik. Durch das Aufdecken der Nuancen, wie spezifische genetische Varianten die Genfunktion und -regulation modulieren, liefert die Feinkartierung wertvolle Einblicke in die regulatorischen Elemente des Genoms. Das Verständnis der Auswirkungen von SNPs auf die Genexpression ist entscheidend für die Identifizierung der molekularen Wege, durch die genetische Variationen zu phänotypischen Ergebnissen beitragen. Dieses detaillierte Wissen ebnet den Weg für die Entwicklung gezielter therapeutischer Strategien und unterstreicht die Bedeutung der Feinkartierung im weiteren Kontext der biomedizinischen Forschung.
Einführung in die SNP-Fine-Mapping
Die feine Kartierung von SNPs ist ein kritischer Schritt, der auf großangelegte Studien folgt. Whole-Genome-SNP-Genotypisierung Studien. Es dient dazu, sich auf spezifische Gene zu konzentrieren, die potenziell mit den interessierenden phänotypischen Merkmalen assoziiert sind. Typischerweise beinhalten Feinkartierungsbemühungen eine reduzierte Anzahl von SNPs, nutzen jedoch eine erheblich größere Stichprobengröße, um die Genauigkeit der Assoziation zu erhöhen.
Nach Abschluss eines umfassenden genomweiten SNP-Screenings und der Identifizierung vorläufiger Zielregionen ist eine Feinabstimmung unerlässlich. Eine effektive Genotypisierung Die Plattform für die feine Kartierung muss eine hohe Anrufrate für die gewählten SNPs aufweisen, auf einem relativ hohen Multiplexniveau arbeiten und langwierige Optimierungsprozesse der Assays vermeiden.
Plattformen, die auf Primerverlängerung und allelspezifischer Ligation basieren, haben sich als wirksam für Feinabstimmungsanwendungen erwiesen. Diese Methoden sind gut geeignet, um die erforderliche Präzision und Effizienz zu erreichen, die notwendig sind, um ursächliche Varianten zu identifizieren, die zur phänotypischen Variation und Krankheitsanfälligkeit beitragen.
Methoden zur Feinkartierung von SNPs
- Heuristische Methoden
Heuristische Ansätze gehören zu den frühesten Techniken, die bei der Feinabstimmung von SNPs eingesetzt werden. Diese Methoden basieren auf empirischen und intuitiven Urteilen, um SNPs anhand ihrer paarweisen Korrelation mit einem führenden SNP zu filtern. SNPs, die eine hohe Korrelation mit dem führenden SNP aufweisen, werden als potenzielle ursächliche Varianten betrachtet. Allerdings sind diese Methoden oft durch willkürliche Schwellenwerte eingeschränkt und es fehlt an einem objektiven Maß für Kausalität.
- Bestrafte Regressionsmodelle
Bestrafte Regressionsmodelle bieten Lösungen für die Probleme der Instabilität und Überanpassung, die häufig bei der Analyse hochdimensionaler Daten auftreten. Durch die Einführung von Strafen während des Prozesses der Likelihood-Maximierung erleichtern diese Modelle die Auswahl von SNPs, die eine starke Assoziation mit dem Merkmal aufweisen, während sie Verzerrungen durch kleinere Effektgrößen minimieren. Zu den bekannten Modellen in dieser Kategorie gehören Lasso, Elastic Net und Minimax Concave Penalty.
- Bayes'sche Methoden
Bayes'sche Methoden bieten einen ausgefeilteren Rahmen, indem sie die posterioren Wahrscheinlichkeiten für SNPs mit nicht null Effekten berechnen. Dieser Ansatz liefert die posterioren Einschlusswahrscheinlichkeiten (PIP), die die Evidenz quantifizieren, die jeden SNP als kausale Variante unterstützt. Bayes'sche Methoden verwenden häufig Techniken der Markov-Ketten-Monte-Carlo (MCMC), um Integrale zu approximieren, wodurch die Auflösung durch Modellvergleiche und die Integration funktionaler Annotationsdaten verbessert wird.
- Integration von Annotationsdaten
Die Integration von Genomannotationsdaten verbessert feine Kartierungsmodelle, indem Informationen zu Genregulation und expressionellen quantitativen Merkmalsloci (eQTL) einbezogen werden. Dieser integrative Ansatz verfeinert die Identifizierung kausaler SNPs, indem sie entsprechend ihrer biologischen Relevanz gewichtet und die a priori Wahrscheinlichkeiten innerhalb von Bayes-Modellen angepasst werden, wodurch die Gesamauflösung verbessert wird.
- Trans-ethnische Feinkartierung
Trans-ethnische feine Kartierung nutzt die genetische Vielfalt verschiedener Populationen, um die Auflösung zu erhöhen. Durch die Kombination GWAS Ergebnisse aus verschiedenen ethnischen Gruppen nutzen diesen Ansatz, um Unterschiede in den Mustern der Kopplungsungleichgewichte (LD) auszunutzen und die Loci der ursächlichen SNPs präziser zu identifizieren. Diese Methode erweist sich als besonders wertvoll in genomischen Regionen, die unterschiedliche genetische Strukturen in verschiedenen Populationen aufweisen.
Unsere SNP-Fine-Mapping-Dienste
Wir bieten mehrere Assaytypen zur Überprüfung von SNP-Markern an, die entdeckt wurden durch RADseq, GBSSNP-Chips oder ähnliche Technologien. Sie umfassen:
-
MassARRAY SNP-Genotypisierung
-
MassARRAY SNP-Genotypisierung verwendet MALDI-TOF-MS, um SNPs durch die Analyse von Massendifferenzen erweiterter DNA-Produkte zu erkennen. Diese Technik bietet hohe Sensitivität, Genauigkeit und Kosteneffizienz. Sie ist besonders vorteilhaft für die Hochdurchsatz-Genotypisierung, erfordert jedoch hochwertige Proben und kann empfindlich auf komplexe Sequenzen in der Umgebung der SNP-Stellen reagieren.
-
-
SNaPshot-Multiplex-System für SNP-Genotypisierung
-
SNaPshot SNP-Genotypisierung verwendet fluoreszenzmarkierte Primer und Kapillarelektrophorese zur Analyse von SNPs. Es ermöglicht die Multiplexierung mehrerer SNPs in einer einzigen Reaktion, was es für Projekte mit mittlerem Durchsatz geeignet macht. Obwohl es spezielle Reagenzien erfordert und kostspieliger sein kann, bietet es hohe Flexibilität und Robustheit bei der Erkennung verschiedener SNPs.
-
-
TaqMan SNP-Genotypisierung
-
TaqMan SNP-Genotypisierung nutzt Echtzeit-PCR mit fluoreszenzmarkierten Sonden, um SNPs präzise anhand der emittierten Fluoreszenz zu identifizieren. Diese Methode ist hochspezifisch und zuverlässig, ideal für großangelegte Studien. Sie bietet eine hervorragende Genauigkeit und ist weithin anerkannt für ihre Benutzerfreundlichkeit, obwohl sie die Synthese von Sonden erfordert und relativ teuer sein kann.
-
Genotypisierungsplattformen, die in der SNP-Fine-Mapping eingesetzt werden, sind sowohl für Uniplex- als auch für Multiplex-Genotypisierung geeignet. Zu den bemerkenswerten Uniplex-Plattformen gehören die TaqMan SNP-Genotypisierung und SNaPshot, während die MassARRAY SNP-Genotypisierung eine zuverlässige Option für die Multiplex-Genotypisierung darstellt. Diese Plattformen zeichnen sich durch ihre hohe Flexibilität und Anpassungsfähigkeit aus. Benutzer können den Durchsatz ausbalancieren, indem sie die Anzahl der analysierten SNPs in Verbindung mit der Probengröße entsprechend den spezifischen Untersuchungsbedürfnissen anpassen. Darüber hinaus bieten sie einen praktischen Vorteil, da fehlgeschlagene SNP-Assays schnell neu gestaltet und erneut bestellt werden können, wodurch die Effizienz und Genauigkeit genetischer Analysen verbessert wird.
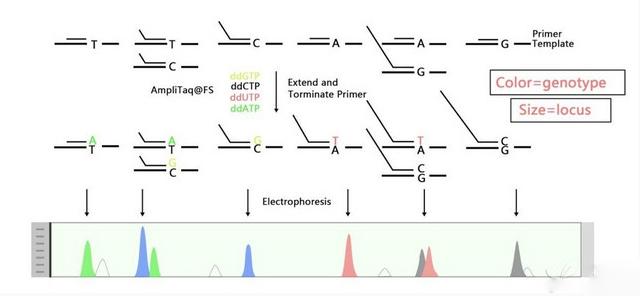 Abbildung 1. Das Prinzipdiagramm von SNaPshot.
Abbildung 1. Das Prinzipdiagramm von SNaPshot.
Vorteile unseres SNP-Fine-Mapping-Services
- Hochauflösende AnalyseCD Genomics bietet präzise SNP-Fine-Mapping mit fortschrittlichen Werkzeugen und umfassenden Datenbanken zur genauen Identifizierung von Varianten an.
- Umfassender methodologischer AnsatzWir wenden verschiedene Techniken an, um trait-assozierte Regionen gründlich zu analysieren und eine zuverlässige Erkennung kausaler SNPs zu gewährleisten.
- Integration von multiethnischen DatenUnsere Dienstleistungen integrieren multiethnische Daten, um die Genauigkeit der SNP-Lokalisierung zu verbessern und eine umfassendere genetische Perspektive zu bieten.
- Expertenunterstützung und maßgeschneiderte LösungenWir bieten persönliche Beratung und hochwertige Ergebnisse durch fachkundige Unterstützung und maßgeschneiderte Lösungen für Ihre Forschungsbedürfnisse.
Unser Expertenteam bietet Beratungsdienste an, um Ihnen zu helfen, die optimale Genotypisierungsstrategie und das experimentelle Design zu bestimmen, die auf Ihre spezifischen Forschungsziele zugeschnitten sind, und dies kosteneffizient und zeitnah.


