
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
CD Genomics bietet seit Jahrzehnten einen genauen und erschwinglichen RNA-Seq (RNA-Sequenzierung) Service an. Wir kombinieren sowohl Illumina (kurze Lesungen) und PacBio (long reads) Plattformen zur Erfassung des Transkriptoms, die eine de novo Assemblierung oder Neusequenzierung ermöglichen für BakterienPflanzen, Tiere und Menschen.
Was ist RNA-Seq?
RNA-Seq, ein entscheidendes Werkzeug, das verwendet wird Next-Generation Sequencing (NGS) Technologien, die darauf ausgelegt sind, detaillierte Karten und Quantifizierungen der Transkriptom, wodurch Informationen wie Gen-Transkriptionsniveaus, die Struktur und Expression von Transkripten, RNA-Modifikationen und nicht-kodierende RNA sowie andere Aspekte aufgedeckt werden. Das Transkriptom, eine umfassende Sammlung aller Transkripte in einer Zelle, bietet wichtige Informationen über Transkriptlevels zu bestimmten Entwicklungsstadien oder physiologischen Zuständen. Das Verständnis des Transkriptoms ist entscheidend, um die funktionalen Elemente des Genoms zu interpretieren sowie biologische Entwicklungen und Krankheiten zu verstehen. Zu den Hauptzielen der Transkriptomik gehören die Katalogisierung aller Transkriptarten; die Bestimmung der transkriptionalen Struktur von Genen; und die Quantifizierung der Expressionsniveaus jedes Transkripts unter verschiedenen Bedingungen.
Durch die Bereitstellung einer unvoreingenommenen hochauflösenden Ansicht globaler Transkriptionsmuster bietet RNA-Seq eine kostengünstige und genaue Methode zur Quantifizierung der Genexpression und zur Analyse der differentiellen Genexpression über mehrere Probengruppen hinweg. Es ermöglicht die Identifizierung neuartiger und zuvor unvorhergesehener Transkripte, unabhängig von einem Referenzgenom, und erleichtert somit die de novo-Assemblierung ununtersuchter Transkriptome. Darüber hinaus ermöglicht es die Entdeckung neuer Genarchitekturen, alternativ gespleißter Isoformen, Genfusionen, SNP/InDel und allelspezifischer Expressionen (ASE).
Vorteile von RNA-Seq
- Quantitative und präzise Messungen von RNA-Molekülen mit einer Auflösung auf Einzel-Basenpaar-Ebene
- Entdeckung neuer Transkripte, Spleißvarianten und Genfusionen
- Bemerkenswerterweise ist diese Strategie auf jede Art anwendbar, unabhängig von der Verfügbarkeit des Referenzgenoms.
- Eine Praxis, die vergleichbare oder sogar niedrigere Kosten im Vergleich zu vielen anderen Methoden bietet.
- Der Ansatz erkennt geschickt verschiedene RNA-Typen, die sich erstrecken über mRNA, miRNA, lncRNA und andere, die einen umfassenden Überblick über die in Zellen oder Geweben vorhandene RNA bieten.
- Bemerkenswert ist die Fähigkeit, mehrere Proben gleichzeitig zu analysieren und dabei effizient umfangreiche Daten zu sammeln – ein Merkmal, das ihren tiefgreifenden Nutzen in der Hochdurchsatz-RNA-Analyse unterstreicht.
RNA-Seq-Entwicklung
Die Sequenzierungstechnologie hat im Laufe der Zeit erhebliche Transformationen und Fortschritte erfahren, insbesondere in den letzten zwei bis drei Jahrzehnten. Zunächst, Sanger-Sequenzierung wurde als die Methode der ersten Generation für die Sequenzierung etabliert. Durch die Nutzung reversibler Terminierungssynthesereaktionen in der binären Chemie ermöglichte die Sanger-Sequenzierung die Bestimmung von Basenfolgen an den DNA-Enden in Übereinstimmung mit der RNA-Sequenz. Die 1990er Jahre markierten bemerkenswerte Fortschritte in der Sequenzierungstechnologie, die mit dem Beginn der Projekte zur Sequenzierung des gesamten Genoms einhergingen. Einführung von Hochdurchsatz-Sequenzierung Plattformen wie die 454-Sequenzierung, Illumina-Sequenzierungund Ion Torrent-Sequenzierung erleichterten die Machbarkeit der RNA-Sequenzierung. Traditionelle RNA-Sequenzierungstechnologien erforderten oft ein erhebliches Volumen an Zellen, um eine zufriedenstellende Menge an RNA für die Sequenzierung zu erhalten, was die Heterogenität zwischen verschiedenen Zellen in den Hintergrund drängte. Im Wesentlichen decken diese Techniken die Merkmale der Genexpression unterschiedlicher Zelltypen und -zustände auf.
Die Anwendungen von RNA-Seq
RNA-Sequenzierung (RNA-seq) ist eine weit verbreitete Technik, die in verschiedenen Bereichen der biologischen und medizinischen Forschung anwendbar ist. Im Folgenden sind mehrere gängige Anwendungen von RNA-seq aufgeführt:
- Genexpressionsanalyse
- Differenzielle Genexpressionsanalyse
- Entdeckung neuartiger Gene
- Analyse des alternativen Spleißens
- Biomarker Entdeckung
- Forschung zu nicht-kodierenden RNAs
- Aufklärung der Genfunktion
- Populationsgenetik und Evolutionsbiologie
In der Tat erweitert sich mit den technologischen Fortschritten der Anwendungsbereich von RNA-Seq unaufhörlich.
Bulk-RNA-Seq vs. zellauflösende Transkriptomik
Bulk-RNA-Seq ermöglicht robuste, kosteneffektive Vergleiche der Genexpression auf Probenebene.
Für Studien, in denen Gewebeheterogenität von Bedeutung ist, kann ein zellauflösender Ansatz helfen, gemischte Signale zu trennen und Veränderungen in der Population oder im Zustand offenzulegen.
Wählen Sie basierend darauf, ob Ihre Frage eine Änderung auf Stichprobenebene oder Einblicke auf zellulärer Ebene betrifft.
| Entscheidungsfaktor | Bulk-RNA-Seq (Probe-Ebene) | Zellauflösende Transkriptomik (zellbasiert / kerngestützt) |
|---|---|---|
| Was Sie messen | Durchschnittliche Expression pro Probe | Ausdruck auf zellulärer Ebene (Heterogenität) |
| Am besten für | DEGs, Wege, globale Veränderungen | Zellpopulationen/-zustände, seltene Signale, Zusammensetzungverschiebungen |
| Typische Eingaben | Breite Kompatibilität (RNA aus Gewebe/Zellen) | Oft benötigt man eine brauchbare Zellvorbereitung. oder kernbasierte Vorbereitung + zusätzliche Qualitätskontrolle |
| Bioinformatik | Standard-QC → Zählungen → DE → Anreicherung | Komplexere QC → Clustering → Annotation → Marker |
| Empfindlichkeit gegenüber Heterogenität | Gemischte Signale können gegensätzliche Veränderungen verschleiern. | Entwickelt, um gemischte Populationen zu trennen. |
| Kosten/Aufwand | Niedriger | Höher |
- Wählen Sie Bulk-RNA-Seq, wenn Ihre zentrale Frage lautet: "Welche Gene/Wegweiser ändern sich zwischen den Bedingungen?"
- Wählen Sie zellauflösende Transkriptomik, wenn Ihre zentrale Frage lautet: "Welche Zellpopulationen treiben die Veränderung voran und wie verändern sich die Zustände?"
Ein mittlerer Weg: Bulk-RNA-Seq + rechnerische Dekonvolution
Wenn geeignete Referenzprofile vorhanden sind, kann die Dekonvolution Trends in der Zellzusammensetzung aus der Gesamtexpression schätzen – nützlich, wenn Experimente mit zellulärer Auflösung nicht durchführbar sind.
RNA-Seq-Workflow
CD Genomics kombiniert sowohl Illumina HiSeq als auch PacBio-Systeme um eine schnelle und präzise RNA-Seq- und bioinformatische Analyse für jede Art bereitzustellen. Unser hochqualifiziertes Expertenteam führt das Qualitätsmanagement durch und befolgt jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten. Der allgemeine Arbeitsablauf für RNA-Seq ist unten skizziert.

Wählen Sie Ihre RNA-Seq-Strategie aus
Verschiedene Forschungsfragen und Probenarten erfordern unterschiedliche RNA-Seq-Strategien. Im Folgenden sind die gängigsten Optionen für die Transkriptom-Profilierung aufgeführt, von gezielter mRNA-Anreicherung bis hin zu umfassenderer Gesamt-RNA-Abdeckung und Long-Read-Isoformauflösung.

mRNA-fokussiertes RNA-Seq (Poly(A)-Anreicherung)
- Am besten fürGenexpressionsquantifizierung, differentielle Expression, Weganalyse, Fusion/Variantenscreening in kodierenden Transkripten
- Überlegungennicht ideal, wenn RNA stark degradiert ist oder wenn nicht-polyadenylierte RNAs im Mittelpunkt der Studie stehen
- Was Sie erhalten werden: Ausdrucksmatrix + DEGs + anreicherungsbereite Ausgaben (optionale Ergänzungen)
Gesamt-RNA-Seq (rRNA-Depletion)
- Am besten fürumfassendere Transkriptomabdeckung einschließlich nicht-kodierender RNAs; Proben, bei denen die Poly(A)-Selektion nicht bevorzugt wird
- Überlegungenerfordert eine robuste rRNA-Depletion und Qualitätskontrolle, um informative Reads sicherzustellen
- Was Sie erhalten werden: Kodierung + nicht-kodierende Ausdrucksausgaben; optionale Entdeckung neuer RNAs/Varianten (projektabhängig)
Vollständige Isoform-Profilierung (Langlese-Option)
- Am besten fürIsoform-Entdeckung, alternatives Splicing, Transkriptannotation, Charakterisierung von Genfusionen
- Wie es verwendet wirdhäufig in Kombination mit Short-Read-RNA-Seq für Quantifizierung + Isoformauflösung
- Was Sie erhalten werden: Volltext-Transkriptmodelle, Isoform-Ebene Annotationen, veröffentlichungsbereite Zusammenfassungsdiagramme (sofern zutreffend)
Erforschen Sie die Sequenzierung von Volltexttranskripten (Iso-Seq)
Gemeinsames DesignKurzzeit-RNA-Seq für robuste Quantifizierung + Langzeit-Isoform-Profiling für Transkriptstruktur und Splicing-Klarheit.
Nicht sicher, welche Strategie zu Ihren Mustern passt? Sprich mit einem Wissenschaftler.
Dienstspezifikationen: Musteranforderungen, Sequenzierung und bioinformatische Analyse
Musteranforderungen und Vorbereitung
|
|
 Klicken |
Sequenzierung
|
 |
Bioinformatische AnalyseWir bieten maßgeschneiderte bioinformatische Analysen an, einschließlich:
|
Analyse-Pipeline
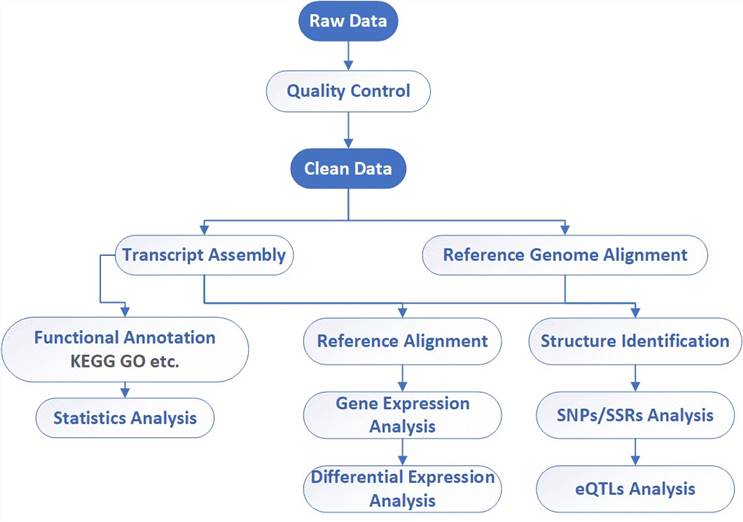
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in RNA-Seq für Ihre Schreibweise (Anpassung)
Entdecken Sie weitere RNA-Sequenzierungsdienste
Neben Transkriptom-RNA-Seq bieten wir spezialisierte RNA-Sequenzierungsassays für nicht-kodierende RNA, Regulationsmechanismen der Translation, Wirt-Mikrobe-Interaktionen sowie für Low-Input- oder extrazelluläre RNA an.
Kleine RNA-Sequenzierung
Profilieren Sie miRNA/siRNA/piRNA-Populationen für Studien zu regulatorischen RNAs.
circRNA-Sequenzierung
Identifizieren und quantifizieren Sie zirkuläre RNAs, um Back-Splicing und regulatorische Mechanismen zu erforschen.
Ribosomen-Profiling (Ribo-seq)
Messen Sie die Translationsaktivität über ribosomen-geschützte Fragmente.
Dual-RNA-Seq
Gleichzeitig die Transkriptome von Wirt und Pathogen erfassen, um Interaktionen zu untersuchen.
Exosomale RNA-Sequenzierung
Profilierung von Vesikel-RNA-Cargo für die Entdeckung von Biomarkern und Signalstudien (RUO).
Ultra-Niedrig-Eingangs-RNA-Seq
Entwickelt für seltene/wertvolle Proben und begrenzte RNA-Mengen.
Degradom-Sequenzierung (PARE)
Kartiere die miRNA-vermittelte Spaltung und validiere Ziele (pflanzenfokussierte Anwendungsfälle üblich).
polyA-Schwanz-Sequenzierung (TAIL-seq)
Quantifizieren Sie die Poly(A)-Schwanzlänge/3'-Endmerkmale, die mit der Stabilität und Translation von mRNA verbunden sind.
Alle Transcriptomics-Dienstleistungen anzeigen →
Brauchen Sie Hilfe bei der Auswahl? Sprich mit einem Wissenschaftler
CD Genomics bietet ein umfassendes RNA-Sequenzierungsdienstleistungspaket an, das die Standardisierung von Proben, den Bibliotheksaufbau, tiefes Sequenzieren, die Qualitätskontrolle der Rohdaten, die Genomassemblierung und maßgeschneiderte bioinformatische Analysen umfasst. Wir können diese Pipeline an Ihr Forschungsinteresse anpassen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenzen:
- Marguerat S, Bähler J. RNA-seq: von der Technologie zur Biologie. Zell- und Molekularbiowissenschaften, 2010, 67: 569-579.
- Hrdlickova R, Toloue M, Tian B. RNA-Seq-Methoden zur Transkriptomanalyse. Wiley Interdisziplinäre Reviews: RNA, 2017, 8(1): e1364.
- Saliba A E, Westermann A J, Gorski S A, et al. Einzelzell-RNA-Sequenzierung: Fortschritte und zukünftige Herausforderungen. Nukleinsäureforschung, 2014, 42(14): 8845-8860.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
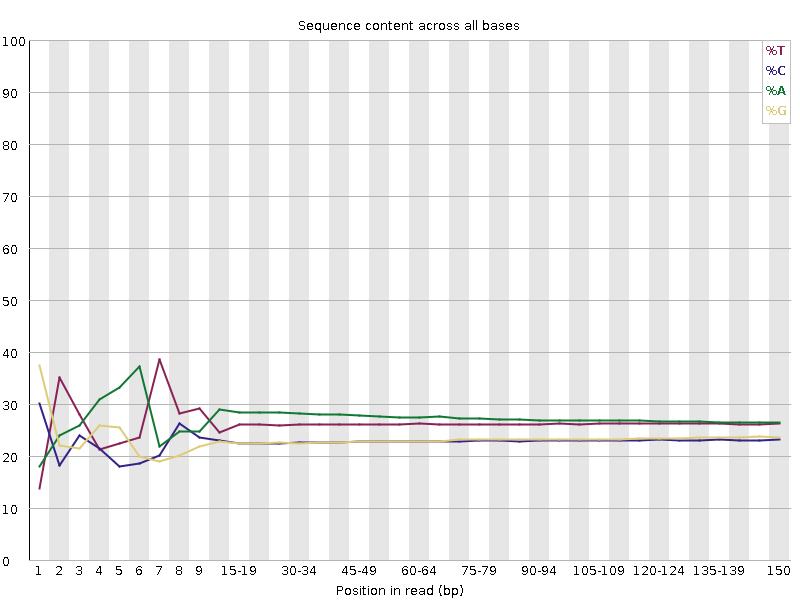
A/T/G/C-Verteilung
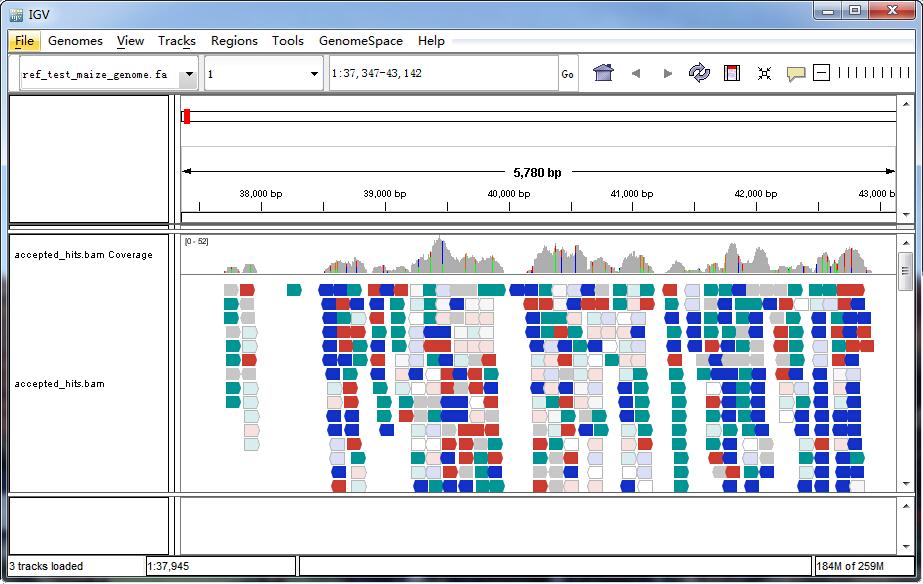
IGV-Browser-Oberfläche
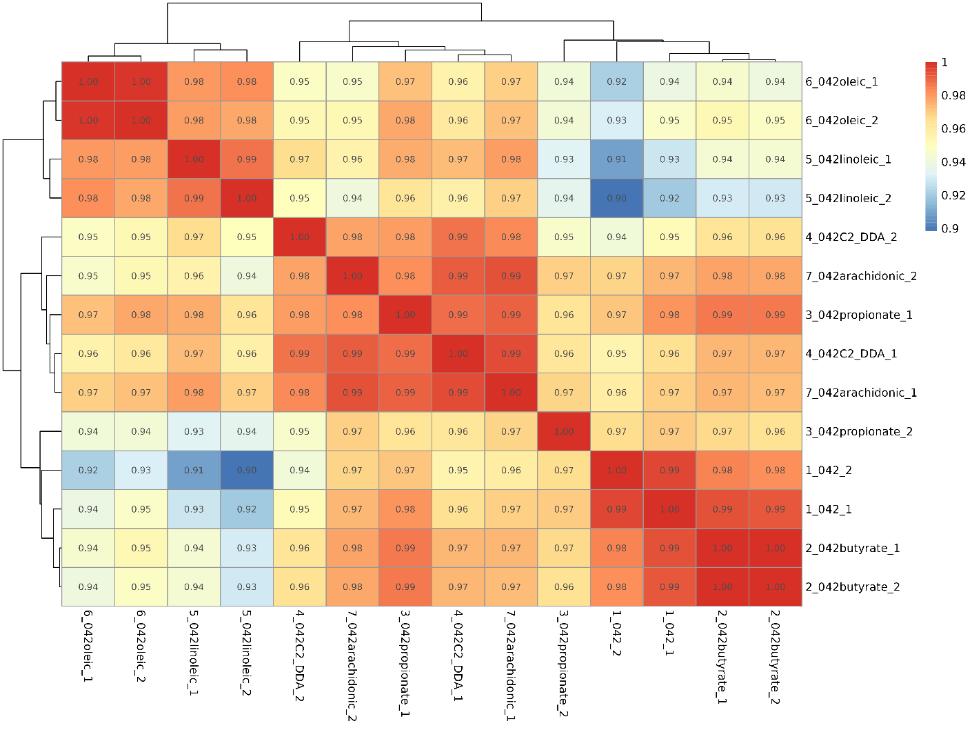
Korrelationsanalyse zwischen Proben
PCA-Score-Diagramm

Venn-Diagramm
Vulkan-Plot
Statistik Ergebnisse der GO-Anmerkung
KEGG-Klassifikation
RNA-Seq häufig gestellte Fragen (FAQs)
1. Wie viele biologische Replikate benötige ich für jede Bedingung?
Wir empfehlen Ihnen, mindestens 3 Replikate pro Probe einzureichen, um das Vertrauen zu erhöhen und experimentelle Fehler zu reduzieren. Beachten Sie, dass dies nur als Richtlinie dient und die endgültige Anzahl der Replikate von Ihnen basierend auf Ihren endgültigen experimentellen Bedingungen festgelegt wird.
2. Welche Vorteile hat NGS gegenüber Microarrays?
Verschiedene Technologien wurden für die Transkriptomanalyse entwickelt, wie zum Beispiel Mikroarray und NGS. Im Vergleich zu Mikroarray hat NGS mehrere Vorteile, darunter:
i. RNA-Seq ist ein empfindliches Werkzeug zur Profilierung der Genexpression. Im Vergleich zu Mikroarray bietet RNA-Seq eine digitale Ablesung, die für die gesamte Genexpression genauer ist.
ii. Mikroarray kann nur begrenzte Informationen zur Genexpression bieten (d.h. die in den Chip integrierten Gene), während NGS einen umfassenderen Ansatz darstellt, der zusätzliche Informationen zu neuartigen Genvarianten und niedrig-abundanten Transkripten liefert.
iii. NGS liefert weitaus reproduzierbarere und zuverlässigere Ergebnisse als Mikroarray. Es ist nicht notwendig, nach RNA-Seq qPCR durchzuführen, während dies ein Standardverfahren für Mikroarray um Ergebnisse zu überprüfen.
3. Wann ist es notwendig, rRNA vor der Sequenzierung zu eliminieren?
Ribosomale RNA (rRNA) macht über 90 % der gesamten RNA aus. Die Durchführung von RNA-Seq ohne Anreicherung für Poly-A oder ohne Depletion von rRNA führt dazu, dass die meisten Reads überwiegend aus rRNA stammen. Zum Beispiel würden weniger als ein Zehntel der Reads nützliche Informationen enthalten. Transkripte aus rRNA-depletierten Transkriptomen werden konventionell als gesamte RNA betrachtet, die umfasst mRNA und nicht-kodierende RNA. Folglich ist die Poly-A-Anreicherung oder die rRNA-Depletion für jede Sequenzierungsplattform unerlässlich.
4. Was ist die konventionelle Pipeline für die RNA-Seq-Datenanalyse?
Die herkömmliche Pipeline für RNA-Seq-Daten umfasst die Qualitätskontrolle der Rohdaten, die Ausrichtung, die Assemblierung, die Genexpressionsprofilierung und andere Analysen.
 Abbildung 1. Übersicht über die RNA-Seq-Datenanalyse (Kukurba und Montgomery 2015).
Abbildung 1. Übersicht über die RNA-Seq-Datenanalyse (Kukurba und Montgomery 2015).
Die Unterscheidung zwischen DNA-Sequenzierung und RNA-Sequenzierung resultiert aus ihren unterschiedlichen analytischen Fähigkeiten. Die DNA-Sequenzierung ermöglicht es uns, die Zusammensetzung der Gene im Genom eines Organismus zu verstehen, ihre Funktionen zu erkennen und ein umfassendes Wissen über intergenetische Beziehungen zu erlangen. Auf der anderen Seite verfeinert die RNA-Sequenzierung unser Verständnis biologischer Mechanismen und Veränderungen, die in Lebensprozessen auftreten. Dies wird erreicht, indem man die Expressionsniveaus, die Vielfalt und die regulatorischen Mechanismen untersucht.
RNA-Sequenzierung zielt speziell auf RNA-Moleküle ab, enthüllt die Struktur und Funktion des Transkriptoms und identifiziert Variationen in der Genexpression. Im Prozess werden RNA-Moleküle in ihre komplementären DNA-Gegenstücke (bekannt als cDNA) transkribiert, die dann sequenziert werden. Zu den häufigen Komponenten der RNA-Sequenzierung gehören in der Regel die vollständige Transkriptomsequenzierung, die Analyse der differentiellen Expression und andere.
Referenz:
- Kukurba K R, Montgomery S B. RNA-Sequenzierung und -Analyse. Cold Spring Harbor Protokolle, 2015(11): pdb. top084970.
RNA-Seq Fallstudien
Transkriptomcharakterisierung durch RNA-Sequenzierung identifiziert eine wesentliche molekulare und klinische Unterteilung bei chronischer lymphatischer Leukämie.
Journal: Genomforschung
Impactfaktor: 11,92
Veröffentlicht: online am 21. November 2013
Zusammenfassung
Die Autoren führten eine tiefgehende RNA-Sequenzierung in verschiedenen Subpopulationen normaler B-Lymphozyten und Zellen der chronischen lymphatischen Leukämie (CLL) aus einer Kohorte von 98 Patienten durch. Sie entdeckten Tausende von transkriptionalen Elementen, die zwischen den CLL- und normalen B-Zellen unterschiedlich exprimiert wurden oder CLL-spezifische Spleißmuster aufwiesen. Sie identifizierten auch eine wesentliche molekulare und klinische Unterteilung in der CLL.
Methoden
- Bioanalyzer 2100
- RNA-seq-Bibliotheken
- cDNA-Bibliotheken
- Illumina-Sequenzierung
- Robuste Multi-Array-Durchschnitt (RMA)
- Gen-, Transkript- und Exonquantifizierungen:
- Flux-Kompensator-Programm
- Ward-Methode
- Die Entfernung 1-r
- Multidimensionale Skalierung
- SPSS Statistics 20.0
- Kaplan-Meier-Methode
- Multivariate Analyse
Ergebnisse
1. Die Genexpressionslandschaft der CLL
Die Autoren fanden 1089 Gene, die zwischen den CLL- und normalen B-Zellen unterschiedlich exprimiert wurden (Tabelle 1). Wie erwartet sind die am stärksten unterschiedlich exprimierten Gene Immunglobuline, bedingt durch die Klonalität der CLL-Zellen. Die Analyse der Signalwege ergab, dass Gene, die an Stoffwechselwegen beteiligt sind, in CLL höher exprimiert wurden, während Gene, die mit dem Spliceosom, dem Proteasom und dem Ribosom in Verbindung stehen, in CLL erheblich herunterreguliert waren.
Tabelle 1. Unterschiede in der Expression und Spleißung zwischen den verschiedenen analysierten Gruppen.
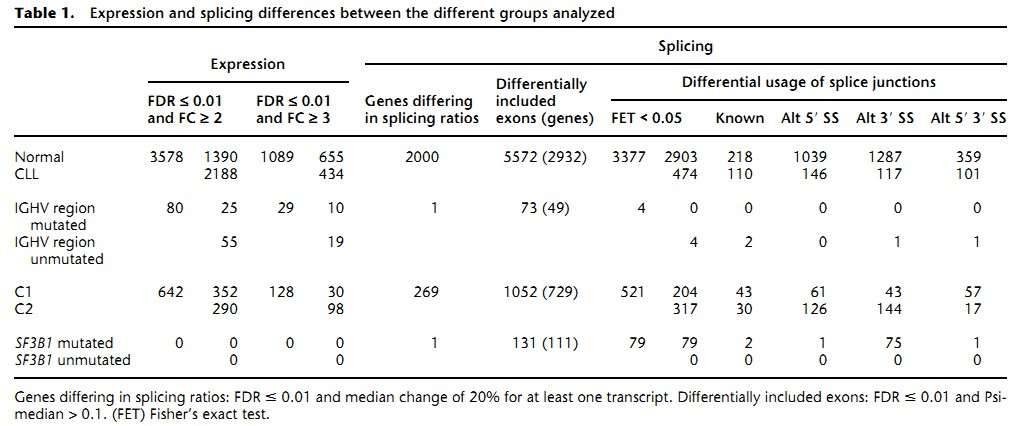
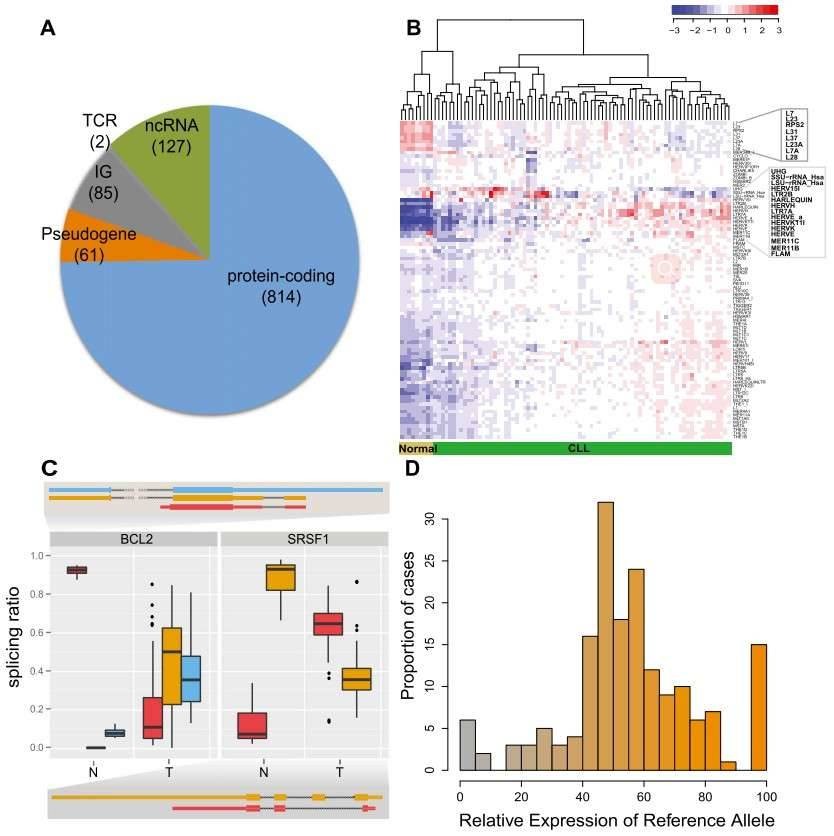 Abbildung 1. Transkriptionale Landschaft der CLL. (A) Das Kodierungspotenzial von differentiell exprimierten Genen zwischen den CLL- und normalen Proben. (B) Normalisierte Expression von transponierbaren Elementen (TEs). (C) Gene mit bedingungsspezifischen Spleißverhältnissen. (D) Allelspezifische Expression somatischer Mutationen.
Abbildung 1. Transkriptionale Landschaft der CLL. (A) Das Kodierungspotenzial von differentiell exprimierten Genen zwischen den CLL- und normalen Proben. (B) Normalisierte Expression von transponierbaren Elementen (TEs). (C) Gene mit bedingungsspezifischen Spleißverhältnissen. (D) Allelspezifische Expression somatischer Mutationen.
2. Die Spleißlandschaft von CLL
Die Autoren identifizierten 2000 Gene mit signifikanten Unterschieden in den relativen Verhältnissen von alternativen Spleißisoformen zwischen CLL- und normalen Zellen, einschließlich Gene mit bekannten alternativen Isoformen als Krebsbiomarker, wie RAC1, CD44 und BCL2L1. Veränderungen im BCR-Signalweg wurden auf der Ebene der Expression und der Spleißung identifiziert (Abbildung 2).
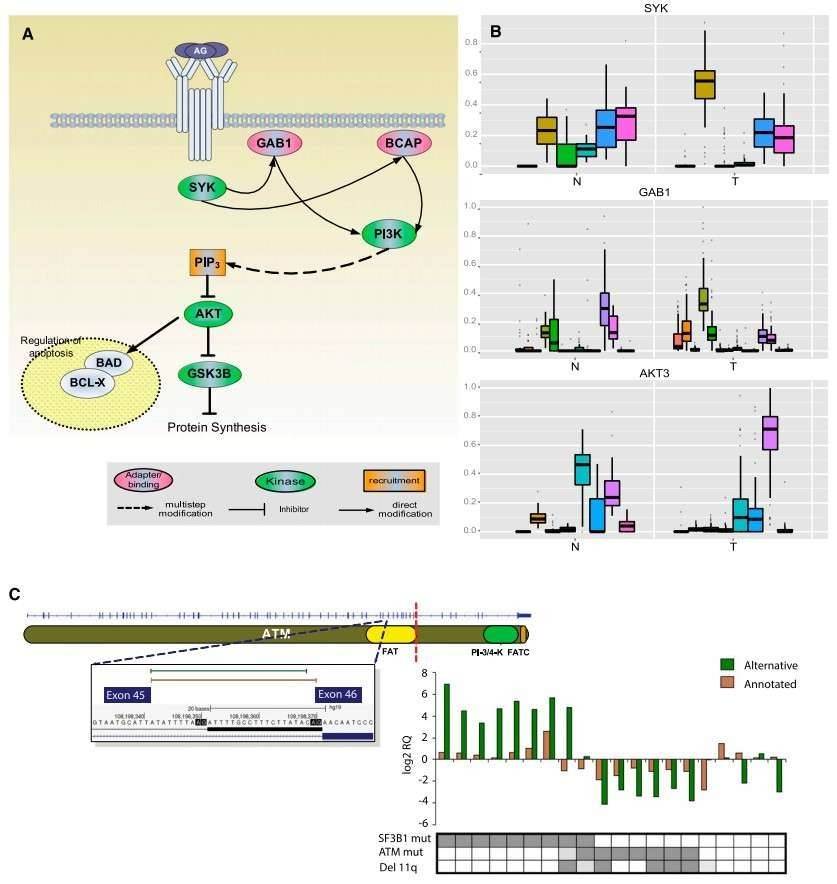 Abbildung 2. Spleißänderungen im BCR-Weg zwischen normalen (N) und Tumor (T) Proben.
Abbildung 2. Spleißänderungen im BCR-Weg zwischen normalen (N) und Tumor (T) Proben.
Transkriptionale Chimären in CLL
Genfusionen, die zu chimären Proteinen führen, stellen einen wichtigen Mechanismus der Karzinogenese dar. Die Autoren identifizierten 122 chimäre Junctions, die ausschließlich in CLL-Zellen mittels RNA-seq-Analyse vorhanden sind. Sie wählten zwei Chimären (die chimäre Junction FCRL2-FCRL3 und die chimäre Junction GAB1-SMARCA5) zur weiteren Validierung durch PCR und Sanger-Sequenzierung aus.
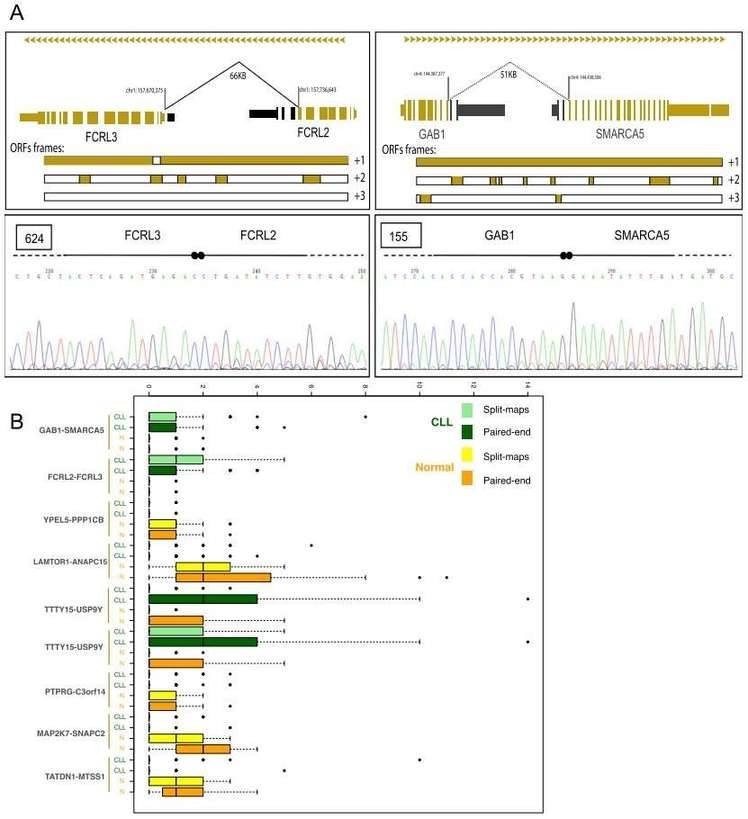 Abbildung 3. Chimäre Verbindungen zwischen FCRL2-FCRL3 und GAB1-SMARCA5.
Abbildung 3. Chimäre Verbindungen zwischen FCRL2-FCRL3 und GAB1-SMARCA5.
4. Identifizierung von zwei wichtigen transkriptionalen CLL-Subgruppen
Die hierarchische Clusteranalyse der RNA-seq-Proben basierend auf der Genexpression trennte deutlich normale Zellen von Tumorproben (Abbildung 4A). Das Clustering offenbarte zwei große, klar definierte Untergruppen innerhalb der CLL-Proben, was durch multidimensionale Skalierung und Hauptkomponentenanalyse weiter unterstützt wurde.
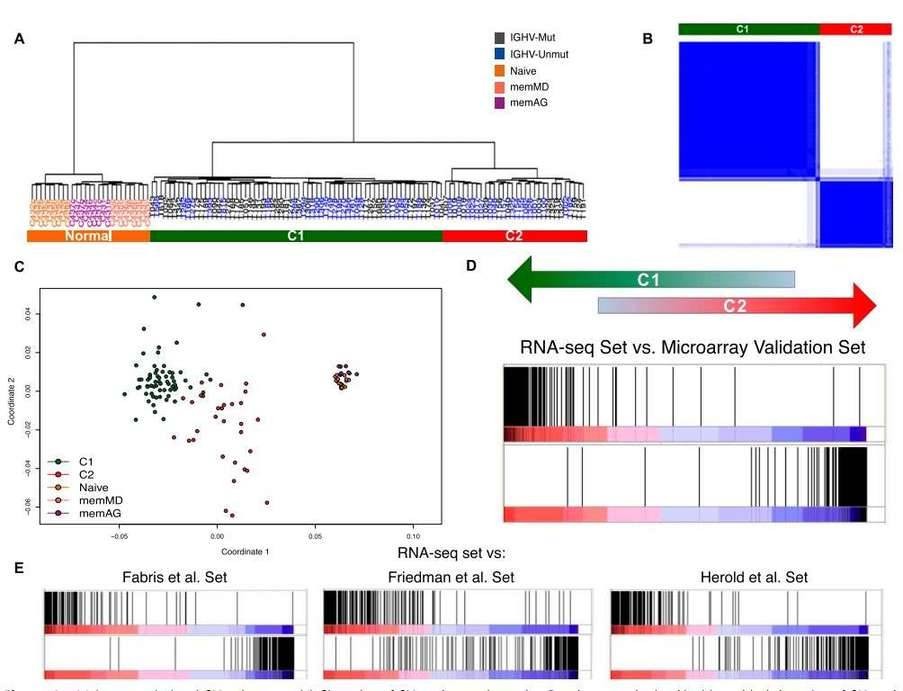 Abbildung 4. Haupttranskriptionale CLL-Subgruppen. (A) Clusterung von CLL- und normalen Proben. (B) Konsenscluster. (C) Multidimensionale Skalierung von CLL- und normalen Proben basierend auf der Genexpression. (D&E) Anreicherungswert-Diagramm.
Abbildung 4. Haupttranskriptionale CLL-Subgruppen. (A) Clusterung von CLL- und normalen Proben. (B) Konsenscluster. (C) Multidimensionale Skalierung von CLL- und normalen Proben basierend auf der Genexpression. (D&E) Anreicherungswert-Diagramm.
5. Klinische Relevanz der C1- und C2-CLL-Gruppen
Die Autoren bewerteten die klinischen Auswirkungen der CLL-Gruppen C1 und C2. Im Vergleich zu C1-Patienten wiesen C2-Patienten eine höhere Häufigkeit von Mutationen in Genen auf, die mit einem ungünstigen Verlauf in Verbindung stehen, und hatten eine höhere Wahrscheinlichkeit, sich im fortgeschrittenen Binet-Stadium zu befinden.
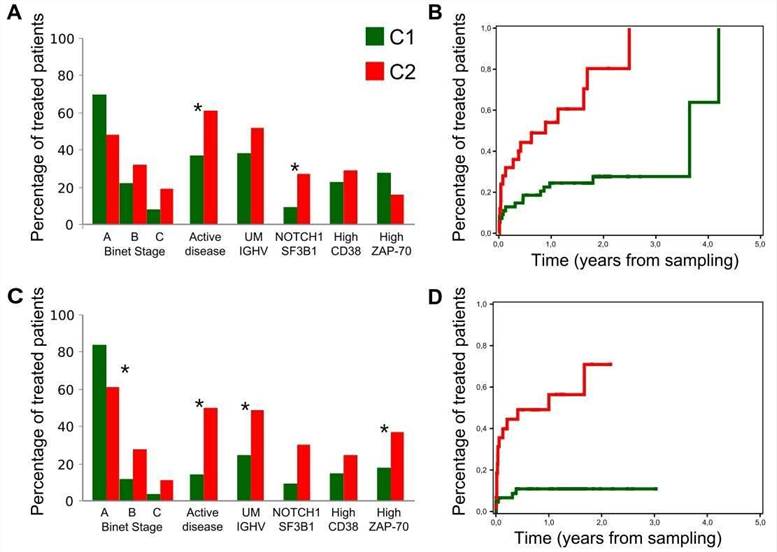 Abbildung 5. Klinisches Verhalten der C1- und C2-Subgruppen.
Abbildung 5. Klinisches Verhalten der C1- und C2-Subgruppen.
Fazit
Wir identifizierten eine differentielle Expression von Tausenden von Transkriptionselementen zwischen CLL- und normalen B-Zellen, einschließlich protein-codierender Gene, nicht-codierender RNAs und Pseudogenen. Darüber hinaus wiesen die meisten Gene CLL-spezifische Spleißmuster auf. Durch die Clusteranalyse von RNA-Sequenzierungsdaten erkannten wir zwei molekulare Subgruppen, C1 und C2, die eng mit klinischen biologischen Merkmalen und der Behandlungsdauer verbunden sind. Nachfolgende Untersuchungen deuteten darauf hin, dass die Aktivierung des B-Zell-Rezeptors (BCR) in der Mikroumgebung der Lymphknoten möglicherweise die Quelle der Unterschiede zwischen C1 und C2 sein könnte.
Referenz:
- Ferreira P G, Jares P, Rico D, et al. Die Charakterisierung des Transkriptoms durch RNA-Sequenzierung identifiziert eine wesentliche molekulare und klinische Unterteilung in der chronischen lymphatischen Leukämie. Genomforschung, 2014, 24(2): 212-226.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Lange nicht-kodierende RNAs könnten den phänotypischen Wechsel von vaskulären glatten Muskelzellen regulieren, indem sie als ceRNA wirken: Implikationen für die Restenose nach Stentimplantation.
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2022
Eine chemosensorähnliche Histidin-Kinase ist in vitro für die Chemotaxis entbehrlich, reguliert jedoch die Virulenz von Borrelia burgdorferi, indem sie die Stabilität von RpoS moduliert.
Journal: PLoS Pathogene
Jahr: 2023
Blut-RNA-Seq-Profiling zeigt eine Gruppe von zirkulären RNAs, die bei gebrechlichen Personen unterschiedlich exprimiert sind.
Zeitschrift: Immunität & Altern
Jahr: 2023
Die gezielte Beeinflussung der CLK2/SRSF9-Spleißachse bei Prostatakrebs führt zu einer verringerten ARV7-Expression.
Journal: Molekulare Onkologie
Jahr: 2024
Hautmikrobielle Gemeinschaft im Zusammenhang mit Erdbeerkrankheiten bei gezüchtetem Regenbogenforelle (Oncorhynchus mykiss Walbaum, 1792)
Journal: Mikroorganismen
Jahr: 2024
Ein recombinationsresistentes Genom für lebend attenuierte und stabile PEDV-Impfstoffe durch die Konstruktion der transkriptionalen Regulierungssequenzen.
Journal: Journal für Virologie
Jahr: 2023
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


