
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Gezielte RNA-Sequenzierung
Gezielte RNA-Sequenzierung stellt einen hochmodernen Service dar, der von führenden Genomik-Anbietern wie CD Genomics angeboten wird. Forscher, die gezielte RNA-seq nutzen, können unvergleichliche Einblicke in Fusionsgene gewinnen und sowohl etablierte als auch zuvor unerkannte Fusionsereignisse auf hohem Detaillierungsgrad erkennen. Solche Fähigkeiten sind in der Onkologie von entscheidender Bedeutung, da sie maßgeschneiderte therapeutische Ansätze ermöglichen und unser Verständnis der Krebsentstehung vertiefen.
Die Einführung der gezielten RNA-Sequenzierung
Die gezielte RNA-Sequenzierung wählt und sequenziert Teiltranskripte von Interesse und erhöht die Abdeckung eines fokussierten Satzes von RNA-Sequenzen. Es handelt sich um eine präzise Methode, die genomische und genexpressionsbezogene Informationen spezifischer Genomregionen erzeugt. Die Analyse spezifischer RNA-Sequenzen ermöglicht RNA-Seq mit hoher Sensitivität und kostengünstigem Zugang zu NGSDurch gezielte Erfassungsanreicherung oder ampliconbasierte Ansätze misst Targeted RNA-Seq Dutzende bis Tausende von Zielen gleichzeitig. Anreicherungsassays sind auch ein Werkzeug, um sowohl bekannte als auch neuartige Genfusionpartner in vielen Probenarten zu identifizieren.
Die gezielte RNA-Sequenzierung liefert qualitative und quantitative Informationen für die Analyse der differentiellen Expression, die Messung der allelspezifischen Expression und die Verifizierung von Genfusionen. Sie kann helfen, Tumorklassifikation und -progression, genetische Erkrankungen und die Reaktion auf RNA-Arzneimittel zu verstehen. Die Sequenzierung des Krebs-Transkriptoms bietet wertvolle Informationen über Veränderungen der Genexpression in Tumoren. Das Profiling von RNA-basierten Biomarkern für die Arzneimittelreaktion trägt dazu bei, multiple medikamentensensible tumorigene Signalwege aufzudecken und die Effizienz sowie die Erfolgsquote des Arzneimittelentwicklungsprozesses zu verbessern.
Vorteile der gezielten RNA-Sequenzierung
- Bietet eine umfassende Analyse der spezifischen Transkripte.
- Wählen Sie validierte Pfade, zell- oder krankheitsspezifische Panels aus.
- Kompatibel mit niedrigqualitativen, formalinfixierten, paraffineingebetteten (FFPE) Proben.
- Schnelle Bearbeitungszeit und höchste Datenqualität
- Stranginformationen zu RNA-Transkripten
- Effektive Transkriptom- und Pfadanalyse
Gezielte RNA-Sequenzierung Arbeitsablauf
Wir bieten integrierte gezielte RNA-Seq-Workflows an, die den gesamten Prozess vereinfachen, von der Bibliotheksvorbereitung bis zur Datenanalyse und biologischen Interpretation.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatische Analyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in der gezielten RNA-Sequenzierung für Ihr Schreiben (Anpassung)
Unterstützt von unserem Team erfahrener Wissenschaftler und fortschrittlicher Technologie bietet CD Genomics gezielte RNA-Sequenzierungsdienste an. Wir wenden strenge Qualitätskontrollmaßnahmen und fortschrittliche bioinformatische Analysen an, um genaue und umfassende Ergebnisse zu gewährleisten. Wenn Sie spezifische Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns für weitere Unterstützung zu kontaktieren.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
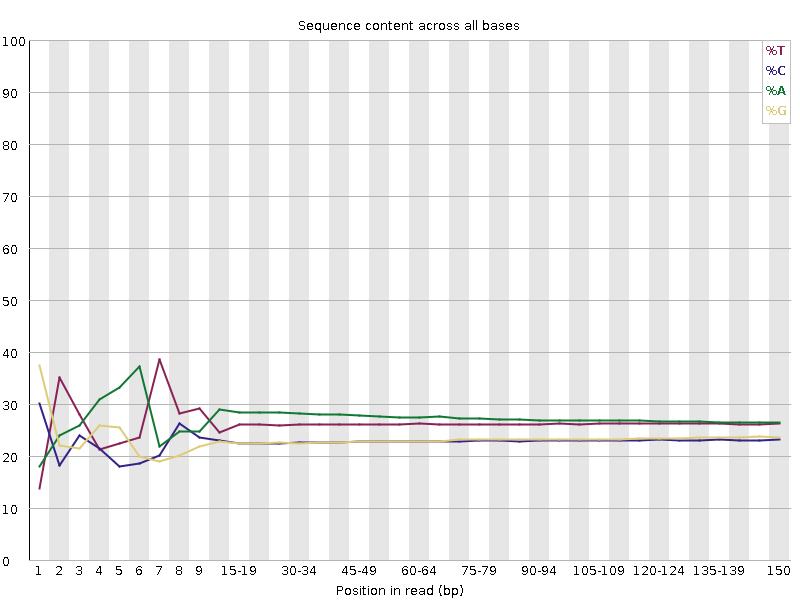
A/T/G/C-Verteilung
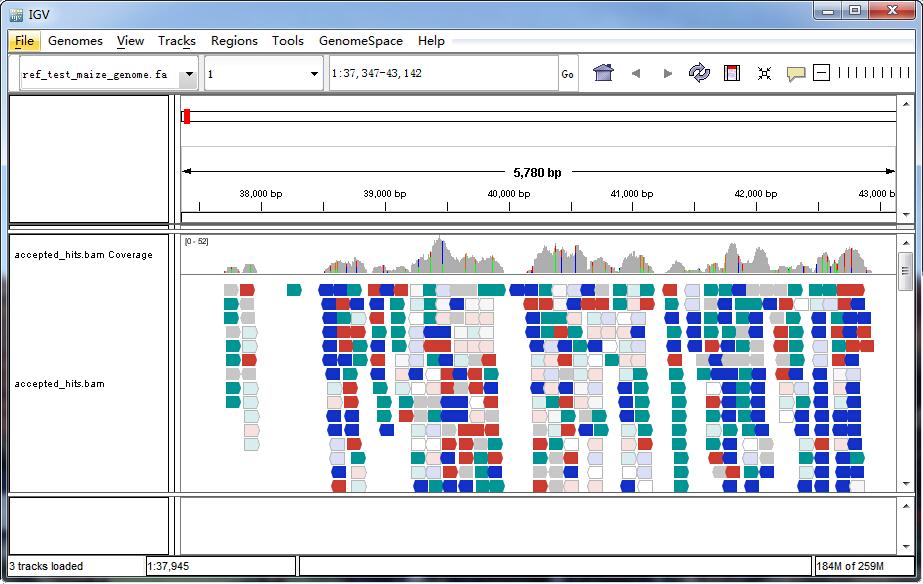
IGV-Browser-Oberfläche
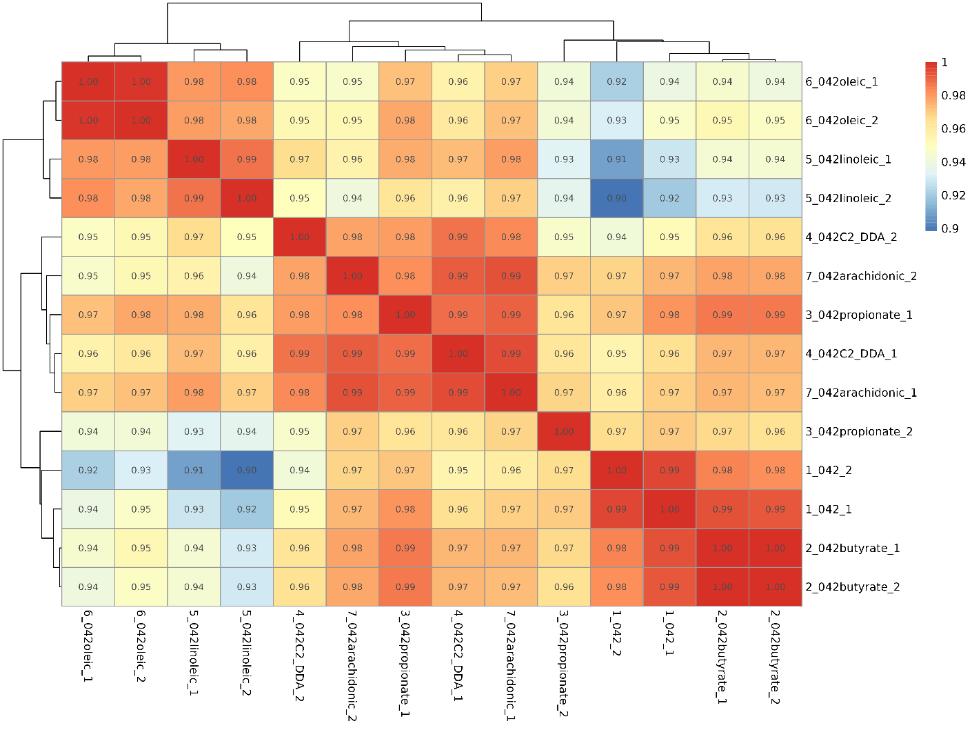
Korrelationsanalyse zwischen Proben
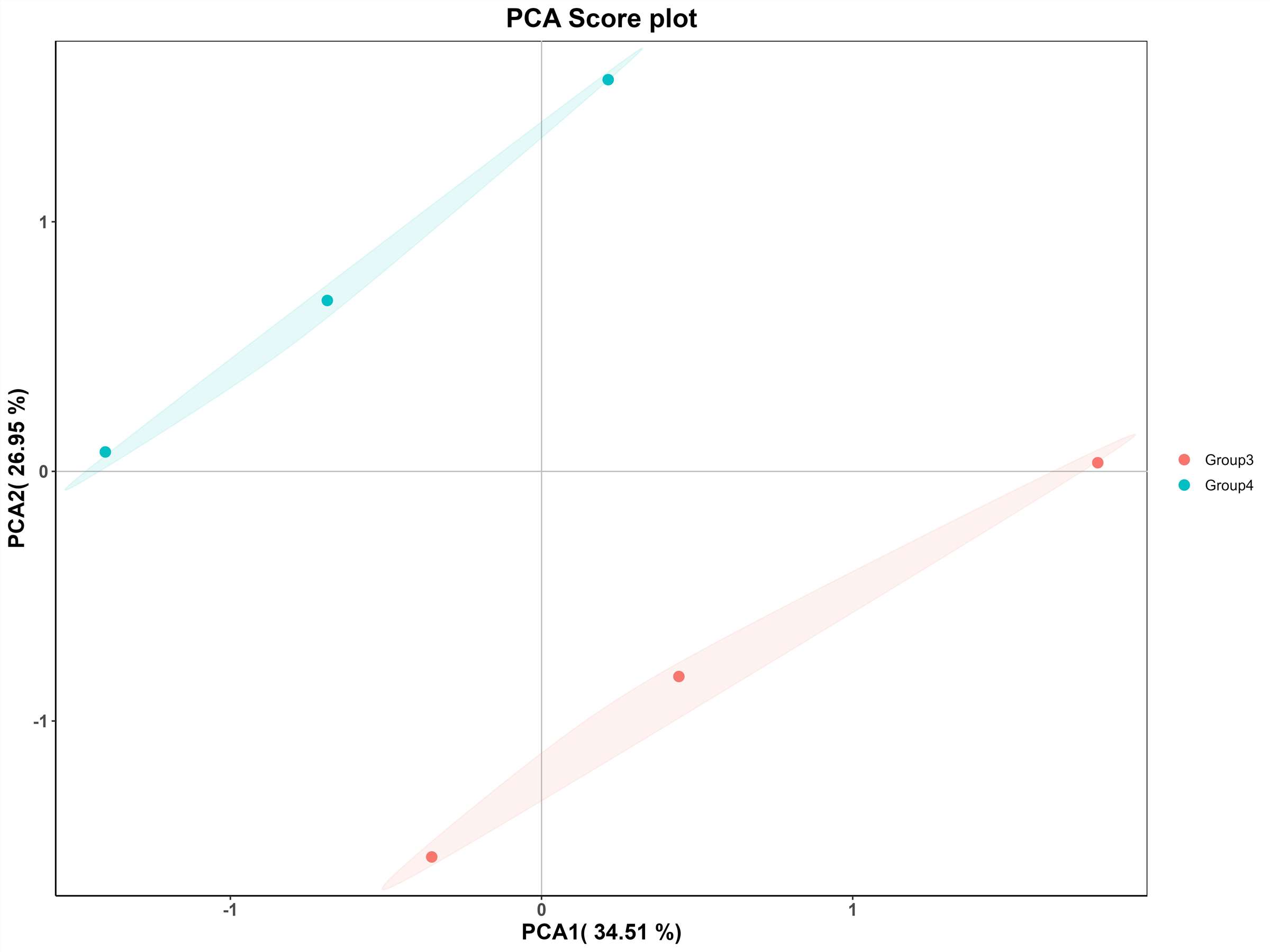
PCA-Score-Diagramm

Venn-Diagramm
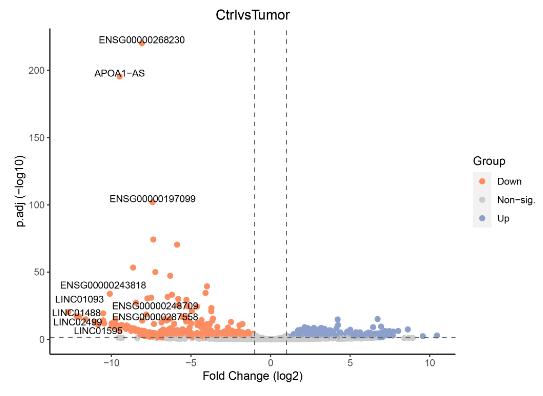
Vulkan-Plot
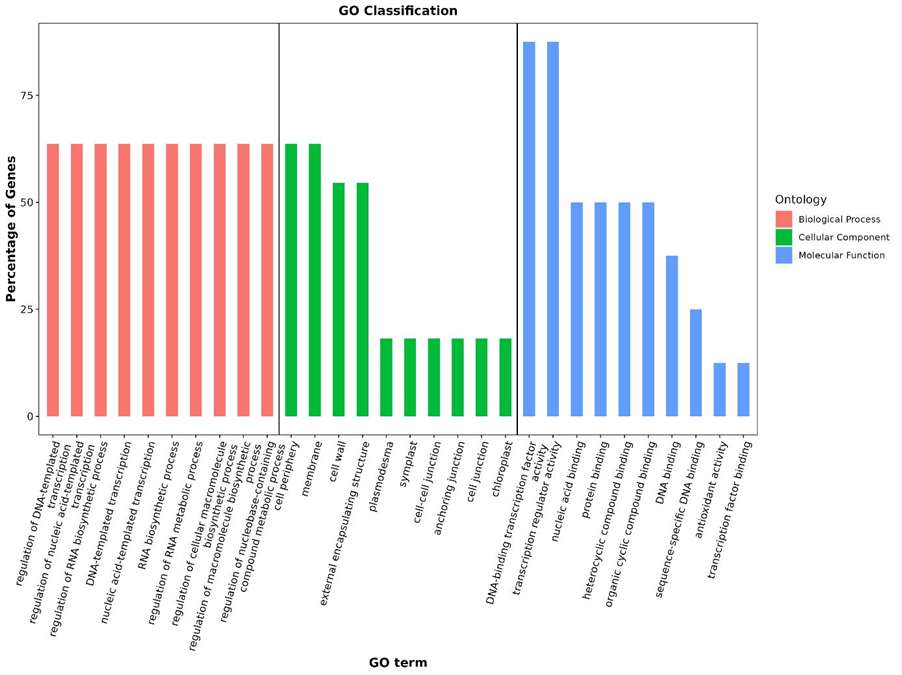
Statistik Ergebnisse der GO-Annotation
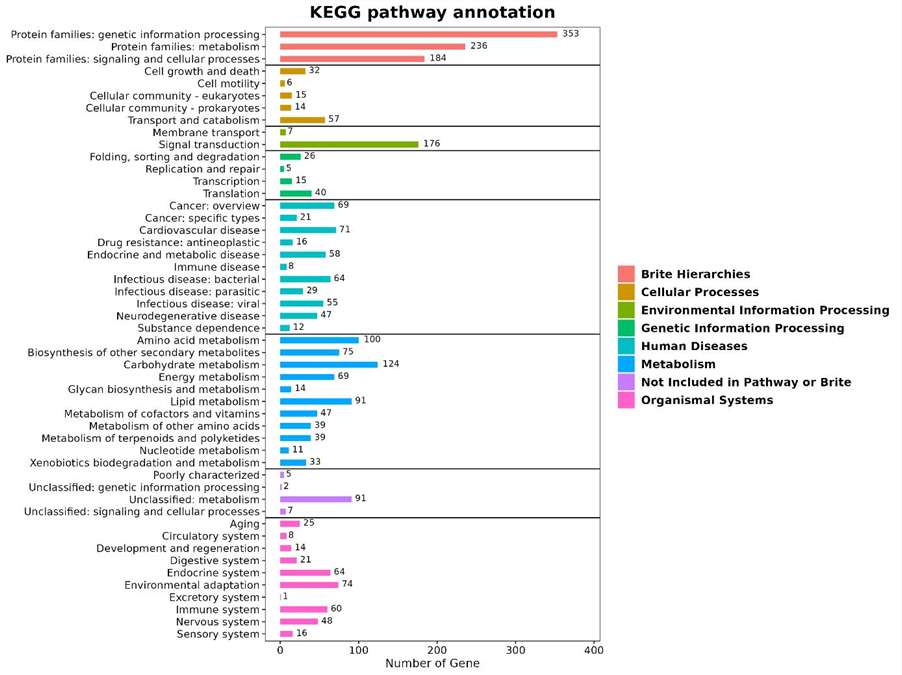
KEGG-Klassifikation
Häufig gestellte Fragen zu gezielter RNA-Sequenzierung
1. Wie unterscheidet sich gezielte RNA-Sequenzierung von der traditionellen gesamten Transkriptom-RNA-Sequenzierung?
Ganz Transkriptom-Sequenzierung ist eine relativ unvoreingenommene Methode zur Erkennung von Fusionstranskripten, die es Forschern ermöglicht, ein breites Spektrum abzudecken und überraschende Ergebnisse zu erzielen. Dieser Ansatz hat jedoch seinen Preis: Ganztranskriptom-Sequenzierung ist nicht nur teuer, sondern hat oft auch eine geringere Sensitivität im Vergleich zu gezielten Sequenzierungsmethoden.
Die gezielte RNA-Sequenzierung hingegen unterscheidet sich darin, dass sie keine Sequenzierungsressourcen erfordert, die über das gesamte Transkriptom verteilt sind, wodurch Forscher sich auf spezifische Transkripte für eine tiefere Sequenzierung konzentrieren können. Wenn sie gut gestaltet sind, können gezielte Methoden die Nachweisempfindlichkeit erheblich erhöhen und gleichzeitig seltene Fusionstranskripte zuverlässig identifizieren.
2. Was sind einige praktische Anwendungen der gezielten RNA-Sequenzierung in der Arzneimittelentwicklung und biologischen Forschung?
Gezielte RNA-Sequenzierung wird in Bereichen wie der Krebsforschung, Neurowissenschaften, Pflanzenbiologie, Mikrobiologie und mehr häufig eingesetzt. Anwendungen umfassen die Identifizierung von Zielen für das Screening von Arzneimitteln, das Studium von Krankheitsmechanismen und die Analyse der Auswirkungen von Umweltstress auf die Genexpression.
3. Wie verbessert die gezielte RNA-Sequenzierung die diagnostischen Möglichkeiten von Fusionsgenen?
Die gezielte RNA-Sequenzierung verstärkt die diagnostischen Fähigkeiten hinsichtlich Fusionsgenen erheblich, indem biotinylierte Oligonukleotid-Sonden eingesetzt werden. Dieses Verfahren reichert RNA-Transkripte an und erleichtert die umfassende Identifizierung zahlreicher Gene, insbesondere solcher, die sporadisch sind oder niedrige Expressionsniveaus aufweisen. Darüber hinaus erkennt diese Strategie gekannte Fusionsgene und beleuchtet bisher unbekannte Begleitfusionsgene.
Gezielte RNA-Seq Fallstudien
Diagnose von Fusionsgenen mittels gezielter RNA-Sequenzierung
Zeitschrift: Naturkommunikationen
Impact-Faktor: 17,694
Veröffentlicht: 27. März 2019
Zusammenfassung
Fusionsgene, die durch chromosomale Umstellungen entstehen, sind entscheidend bei Krebs und tragen zu etwa 20 % der Fälle bei, mit unterschiedlicher Häufigkeit in den verschiedenen Krebsarten. Eine schnelle und genaue Erkennung ist entscheidend für eine präzise Diagnose und Behandlung. Aktuelle Methoden wie FISH und RT-PCR sind zwar sensitiv, übersehen jedoch häufig neuartige Fusionspartner und komplexe Umstellungen, was zu Fehldiagnosen bei hämatologischen Krebserkrankungen führt. RNA-Sequenzierung bietet umfassende Überwachung, hat jedoch Schwierigkeiten mit der Sensitivität für gering exprimierte Fusionsgene. Zielgerichtete RNA-Sequenzierung unter Verwendung biotinylierter Sonden verbessert die Erkennung seltener Transkripte und zeigt vielversprechende Ansätze für präzise Fusionsgen-Diagnosen bei soliden Tumoren und Lungenkrebs.
Materialien & Methoden
Probenvorbereitung
Periphere Blutproben
RNA-Extraktion
Sequenzierung
Bibliotheksbau
cDNA-Erfassung
Illumina HiSeq 2500 v4.0 Plattform
RT-PCR und Sanger-Sequenzierung
Fusionsdetektion
Änderung der Genabdeckung
Transkriptom-Assemblierung
Analyse von Immunrezeptoren
Ergebnisse
Die gezielte RNA-Sequenzierung wurde zur Diagnose von Fusionsgenen in Zelllinien und Tumorproben von Patienten validiert. Sie identifizierte erfolgreich bekannte Fusionsgene wie ROS1 und ALK in Biopsien von Lungenkrebs und bestimmte sowohl die Genpartner als auch die Fusionsjunctionen. In einer breiteren klinischen Kohorte von 72 Proben, einschließlich solider Tumoren und hämatologischer Malignitäten, entdeckte die gezielte RNA-Sequenzierung in 76 % der Fälle Fusionsgene und übertraf damit traditionelle Methoden. Die Reproduzierbarkeit war hoch über die Replikatproben hinweg, was ihre Zuverlässigkeit für den klinischen Einsatz unter Beweis stellte. Die Methode entdeckte auch neuartige Fusionsgene und Isoformen, was ihr Potenzial zur Unterstützung einer personalisierten Krebsbehandlung hervorhebt.
 Abb. 1. Fusionidentifikation in klinischen Kohortenproben.
Abb. 1. Fusionidentifikation in klinischen Kohortenproben.
Targetiertes RNA-Seq identifiziert nicht nur Fusionsgene, sondern quantifiziert auch die Genexpression über die erfassten Gene hinweg. Es zeigt unterschiedliche Expressionsmuster, wie die hohe Expression von Fusionsgenen wie EZR-ROS1, und erkennt Fälle, in denen Promoterfusionen Veränderungen der Genexpression antreiben, wie bei ACSL3-ETV1. Trotz seiner Sensitivität kann das zielgerichtete RNA-Seq Fälle von Promoterfusionen übersehen, wie in einer Sarkomprobe mit erhöhter ROS1-Expression. Darüber hinaus bietet es Einblicke in die Expression von Marker-Genen, wie GATA2 in AML und CML, die mit der Prognose korrelieren.
 Abb. 2. Vielfalt der Fusionsjunctionen und Genexpression.
Abb. 2. Vielfalt der Fusionsjunctionen und Genexpression.
Die Blutuntersuchung der Autoren zielte auf V-, J- und C-Exons an IG/TCR-Rezeptorloci ab, um Fusionsgene wie IGH-MYC und IGH-BCL6 bei Lymphom-Patienten zu identifizieren. Diese Sonden erfassten auch RNA-Transkripte von diesen Loci, was eine Profilierung des Immunrepertoires ermöglichte. Die Analyse von Zelllinien zeigte dominante Klonotypen, die mit klonalen Populationen übereinstimmten, während Patientenmuster vielfältige Klonotypen von Immunrezeptoren aufwiesen, insbesondere in der B-Zell-Reifung im Knochenmark, wobei bemerkenswerte Expansionen möglicherweise auf maligne klonale Populationen in einigen Fällen hinwiesen.
 Abb. 3. Neuartige Erkenntnisse in der transkriptomischen Analyse.
Abb. 3. Neuartige Erkenntnisse in der transkriptomischen Analyse.
Fazit
Zusammenfassend sind chromosomale Translokationen, die Fusionsgene erzeugen, entscheidend für Krebs und erfordern eine präzise Diagnose für eine effektive Behandlung. Im Gegensatz zu traditionellen Methoden bietet gezielte RNA-Sequenzierung eine umfassende Erkennung bekannter und neuer Fusionsgene über Hunderte von Genen hinweg, was eine schnellere und genauere Diagnose ermöglicht. Trotz ihrer Hochdurchsatzfähigkeit erfordert die gezielte RNA-Sequenzierung eine sorgfältige bioinformatische Überwachung, um falsch-positive Ergebnisse zu minimieren. Ihre Fähigkeit, komplexe Umstellungen aufzulösen und alternative Isoformen zu erkennen, verbessert die prognostischen Einblicke und die Behandlungsplanung, während ergänzende Analysen wie die Genexpressionsprofilierung und das Klonotyping von Immunrezeptoren die diagnostische Präzision weiter verfeinern. Letztendlich ist die gezielte RNA-Sequenzierung bereit, die Diagnostik von Fusionsgenen zu revolutionieren und bietet eine breitere klinische Anwendbarkeit sowie tiefere Einblicke in die Krebsbiologie.
Referenz
- Heyer EE, Deveson IW, Wooi D, et al. Diagnose von Fusionsgenen mittels gezielter RNA-Sequenzierung. Naturkommunikationen2019, 27;10(1):1-2.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Chaperon-vermittelte Autophagie steuert proteomische und transkriptomische Wege, um die Aktivität von Gliom-Stammzellen aufrechtzuerhalten.
Zeitschrift: Krebsforschung
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 bei Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikationen vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzelligkeit und die Tumorbildung im Blinddarm zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neonatalen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


