
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Pan-Genom
Die Einführung der Pan-Genom-Sequenzierung
Das Konzept des Pan-Genoms umfasst die Gesamtheit der Gene, die in allen Stämmen einer bestimmten Art vorhanden sind, und bietet somit eine umfassende Perspektive auf die genetische Vielfalt, die die Grenzen individueller Genomsequenzen überschreitet. Es umfasst zwei Hauptkomponenten:
- Kern-Genom: Dieses Segment umfasst Gene, die in allen Stämmen der Art ubiquitär vorhanden sind. Diese Kern-Gene sind typischerweise mit grundlegenden biologischen Funktionen und primären phänotypischen Merkmalen assoziiert, wodurch die evolutionäre Stabilität der Art widergespiegelt wird. Zum Beispiel in Escherichia coliKern-Gene umfassen solche, die an wesentlichen zellulären Prozessen wie DNA-Replikation und Transkription beteiligt sind.
- Variabler (Zubehör/Entbehrlicher) Genom: Im Gegensatz dazu umfasst dieses Segment Gene, die nur in einer Teilmenge von Stämmen oder Individuen innerhalb der Art vorkommen. Diese Zubehörgene kodieren häufig für spezifische Anpassungen oder einzigartige Merkmale, einschließlich Faktoren wie Antibiotikaresistenz oder Virulenz. Zum Beispiel tragen in Streptococcus pneumoniae Zubehörgene zu Variationen in der Pathogenität und den Resistenzprofilen verschiedener Stämme bei.
Durch die umfassende Untersuchung des Pan-Genoms können Forscher Einblicke in das gesamte Spektrum genetischer Variabilität innerhalb einer Art gewinnen, was ein tieferes Verständnis ihrer evolutionären Dynamik, funktionalen Anpassungen und potenziellen Reaktionen auf Umweltbelastungen ermöglicht.
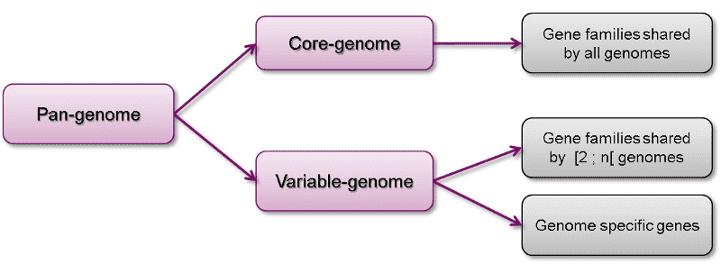 Abbildung 1. Zusammensetzung eines Pan-Genoms
Abbildung 1. Zusammensetzung eines Pan-Genoms
Die Verfolgung der Pan-Genom-Sequenzierung beinhaltet die Nutzung von Hochdurchsatz-Sequenzierung Technologien neben anspruchsvollen bioinformatischen Werkzeugen. Dieser Ansatz umfasst den sorgfältigen Aufbau von Sequenzierungsbibliotheken, gefolgt von einer umfassenden Sequenzierung von Individuen, Unterarten oder Linien innerhalb einer Art. Die anschließende Zusammenstellung dieser Sequenzen führt zur Entwicklung einer Pan-Genom-Karte. Diese Karte dient dazu, das genetische Repository der Art zu bereichern und ermöglicht die Untersuchung wichtiger biologischer Fragen, was erheblich zu unserem Verständnis von genetischer Vielfalt und evolutionären Prozessen beiträgt.
Warum ist die Pan-Genom-Forschung unerlässlich?
Im umfangreichen Verlauf der Evolution, beeinflusst von geografischen und umweltbedingten Faktoren, entwickeln einzelne Organismen hochgradig einzigartige genetische Merkmale. Ein einzelnes Referenzgenom reicht nicht aus, um die vollständigen genetischen Informationen einer gesamten Art zu erfassen. Mit anderen Worten kann die ausschließliche Abhängigkeit von einem einzigen Referenzgenom für Studien zur genetischen Domestikation und Variation dazu führen, dass bedeutende genomische Inhalte verloren gehen, da viele einzigartige Sequenzen im Referenzgenom nicht vertreten sind.
Darüber hinaus hat der sinkende Preis für die Genomsequenzierung die Forschung zu Pan-Genomen in den letzten Jahren zunehmend machbar gemacht. Diese Kostenreduktion erleichtert die Erforschung der genetischen Vielfalt in einer Tiefe und im Umfang, die zuvor unerreichbar war. Folglich ist das Studium von Pan-Genomen zu einem aufstrebenden Feld geworden, das tiefgreifende Einblicke in die Komplexität genetischer Informationen und das Anpassungspotenzial von Arten bietet.
Unterschied zwischen dem Pan-Genom und dem gesamten Genom
Das Konzept des gesamten Genoms bezieht sich auf den vollständigen Satz genetischen Materials, der innerhalb eines einzelnen Individuums oder Stammes gefunden wird, und wird typischerweise als Referenz in der Genomforschung verwendet. Im Gegensatz dazu vereint das Pan-Genom genetische Informationen aus mehreren Individuen oder Stämmen und bietet eine ganzheitlichere und umfassendere genetische Landschaft der Art.
Wesentliche Unterschiede
- UmfangDas gesamte Genom umreißt die genetische Architektur eines einzelnen Stammes. Im Gegensatz dazu umfasst das Pan-Genom alle genetischen Varianten, die über mehrere Stämme hinweg vorhanden sind, und erfasst somit ein breiteres und umfassenderes Spektrum genetischer Vielfalt.
- AuflösungDie Pan-Genom-Analyse beleuchtet genetische Variationen, die möglicherweise unentdeckt bleiben, wenn man sich ausschließlich auf ein einzelnes Referenzgenom stützt. Dies umfasst einzigartige accessory Gene, die stammspezifische Eigenschaften und Anpassungen vorantreiben.
Zum Beispiel die Untersuchung des Pan-Genoms von Mycobacterium tuberculosis zeigt erhebliche genetische Variabilität zwischen verschiedenen Stämmen, ein Faktor von größter Bedeutung für das Verständnis der Mechanismen der Arzneimittelresistenz und für die Entwicklung effektiver therapeutischer Interventionen.
Vorteile des Pan-Genoms
- Bereichern Sie die Genominformationen der Art durch Sequenzierung der Unterarten und Individuen.
- Schnell Gene oder strukturelle Variationen von Genen finden, die mit wichtigen Merkmalen in Verbindung stehen, basierend auf der Untersuchung des variablen Genoms.
- Untersuchen Sie die Unterschiede innerhalb von Arten aus der Perspektive einzigartiger Genfolgen.
- Kleine Bevölkerung, kosteneffizient und zeiteffizient
Anwendungen des Pan-Genoms
- Artenentwicklung und PhylogenetikHilft bei der Nachverfolgung evolutionärer Beziehungen und genetischer Vielfalt zwischen Arten.
- Merkmalsentdeckung und ZuchtIdentifiziert Gene, die mit wünschenswerten Eigenschaften für eine verbesserte Pflanzenzüchtung verbunden sind.
- Pathogenforschung: Enthüllt genetische Variationen in Krankheitserregern, die mit Virulenz und Resistenz in Verbindung stehen, und informiert über Kontrollmaßnahmen.
- Ökologische und UmweltstudienBietet Einblicke in die Anpassungen von Arten an verschiedene Umgebungen und ökologische Nischen.
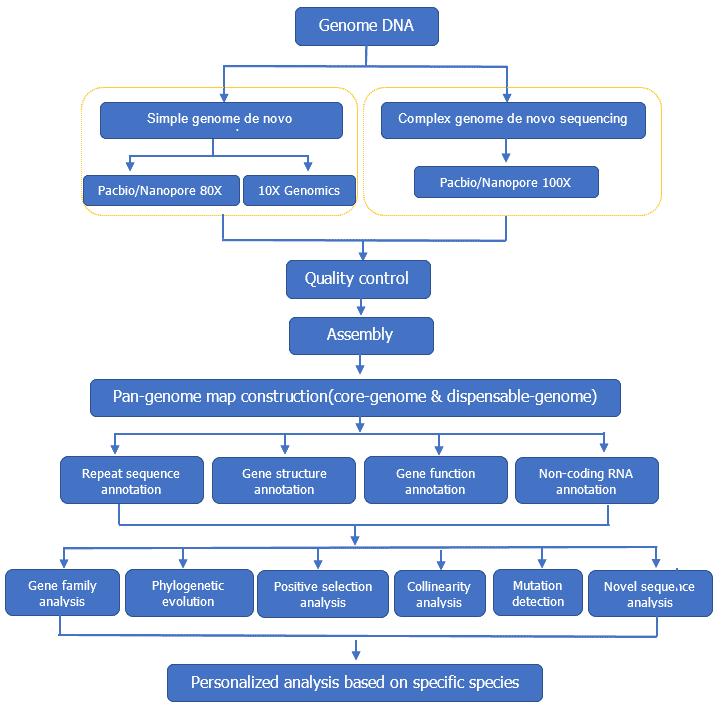
Pan-Genom-Workflow

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details im Pan-Genom für Ihre Schreibanpassung.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
Referenzen
- Liu Y, Du H, Li P, et al. Pan-Genom von wilden und kultivierten Sojabohnen. Zelle2020, 182(1):162-76.
- Chen J, Liu Y, Liu M, et al. Pangenom-Analyse zeigt genomische Variationen im Zusammenhang mit Domestikationsmerkmalen bei Hirse. Naturwissenschaften Genetik2023, 55(12):2243-54.
Pan-Genom FAQs
1. Wie viele Proben sind für die Pan-Genom-Sequenzierung erforderlich?
Mindestens zwei Individuen oder Unterarten sind erforderlich, um eine Pan-Genom-Analyse zu beginnen. Um jedoch ein umfassenderes Verständnis der genetischen Vielfalt innerhalb einer Art zu erreichen, sollte eine größere Anzahl von Proben in Betracht gezogen werden. Die Einbeziehung zusätzlicher Proben verbessert erheblich die Auflösung der genetischen Variation und führt zu einer umfassenderen Pan-Genom-Karte.
2. Ist ein Referenzgenom für die Pan-Genom-Analyse erforderlich?
Die Nutzung eines Referenzgenoms in der Pan-Genom-Analyse ist nicht strikt erforderlich, kann jedoch von Vorteil sein. Ein Referenzgenom dient als anfänglicher Rahmen für die Genannotierung und den vergleichenden Vergleich. Dennoch besteht das Hauptziel von Pan-Genom-Studien darin, genetische Variationen aufzudecken, die den Rahmen eines einzelnen Referenzgenoms überschreiten. Daher ist die Pan-Genom-Analyse darauf ausgelegt, genetische Vielfalt zu identifizieren und zu integrieren, die über das hinausgeht, was durch ein einzelnes Referenzgenom dargestellt wird, und somit eine umfassende Erfassung der genomischen Landschaft der Art sicherzustellen.
3. Welche Arten von Analysen werden in standardmäßigen Pan-Genom-Diensten durchgeführt?
Pan-Genom-Assemblierung: Beinhaltet die Analyse des GC-Gehalts, die Analyse der Sequierungstiefe und den Aufbau von Super-Scaffolds unter Verwendung von Referenzgenomen.
Pan-Genom-Strukturanalyse: Beinhaltet die Vorhersage von Kern- und Accessory-Genomen, die Annotation von Wiederholungssequenzen und die Identifizierung von nicht-kodierenden RNAs.
Pan-Genom Fallstudien
Pangenomanalysen zeigen den Einfluss von transponierbaren Elementen und Ploidie auf die Evolution von Kartoffelarten.
Journal: Mitteilungen der Nationalen Akademie der Wissenschaften
Impact-Faktor: 12,779
Veröffentlicht: 24. Juli 2023
Hintergrund
KartoffelKartoffel) ist eine wichtige globale Kulturpflanze mit reicher genetischer Vielfalt. Ursprünglich aus den Anden, umfasst sie mehrere Ploidie-Ebenen. Diese Studie präsentiert das umfassendste Kartoffel-Pan-Genom, das Sequenzen von 296 Zugängen kombiniert und 132.355 Pangenen identifiziert. Die Analyse hebt die genetische Vielfalt, Anpassung und evolutionären Beziehungen innerhalb der Solanum-Sektion hervor. Petota.
Materialien & Methoden
Probenvorbereitung
- Kartoffel
- Genomsequenzen von Kartoffelzugängen
Methode
- Sequenzierungsdaten herunterladen
- Konstruktion des Pangenoms
- De novo Versammlung
- Ausrichtung
- Annotation des Pangenoms
- PAV-Analyse
- Analyse der Unterschiede in der Genfrequenz
Ergebnisse
1. Das Pangenom von Nachtschatten Abschnitt Petota
Das Pangenom umfasst 296 Kartoffelproben mit unterschiedlichen Ursprüngen und Ploidieebenen, einschließlich 154 Gbp neuer und öffentlicher Assemblierungen, was zu 514.888 Contigs und 132.355 Genmodellen führt.
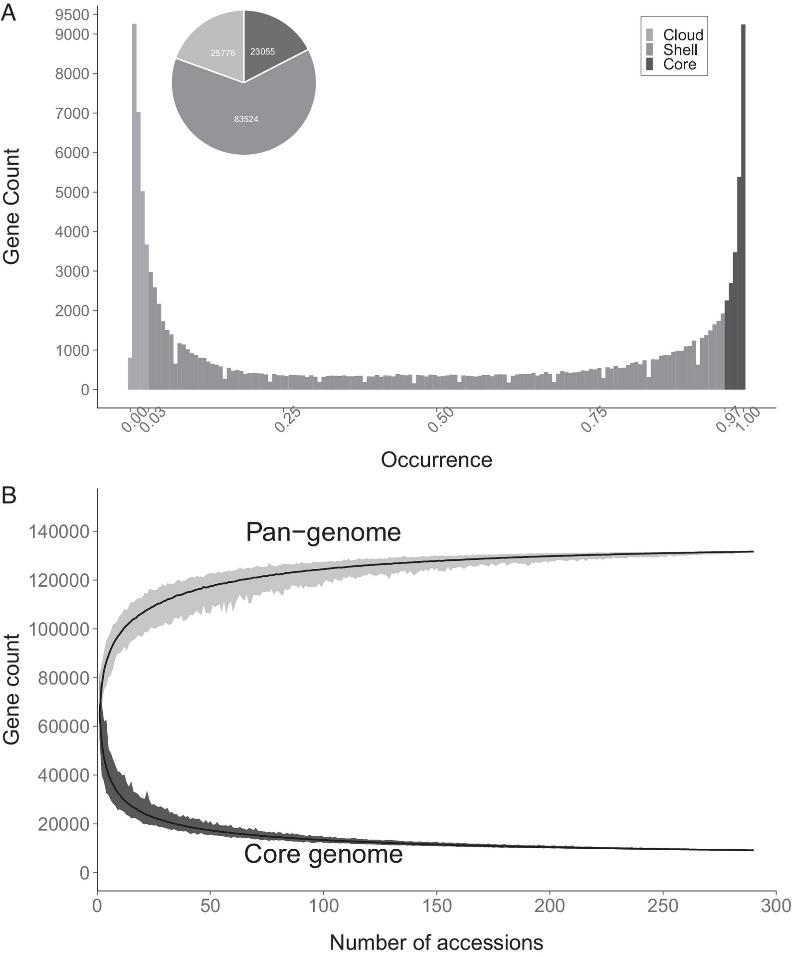 Abb. 1. Das Nachtschatten Abschnitt Petota Pangenom.
Abb. 1. Das Nachtschatten Abschnitt Petota Pangenom.
2. PAV-Variation in protein-kodierenden Genen
Die Variation in protein-codierenden Genen zwischen Proben wird durch Ploidie und Domestikation beeinflusst, was die Genanzahl und Selektion betrifft.
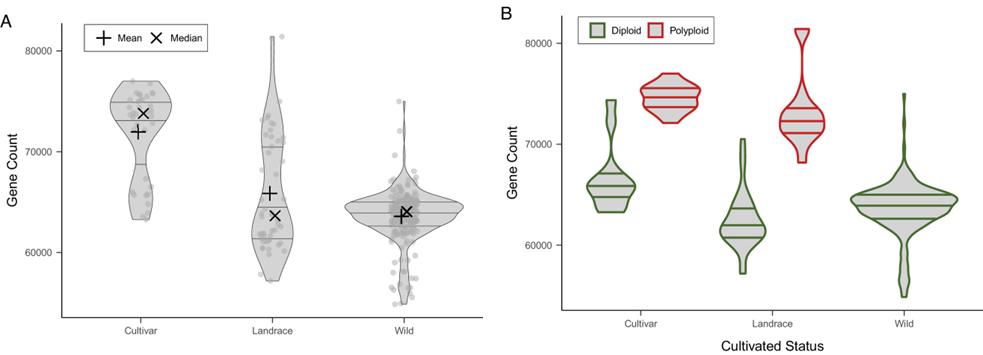 Abb. 2. Geninhalt der in den Proben enthaltenen Zugänge Nachtschatten Abschnitt Petota Pangenom basierend auf PAV.
Abb. 2. Geninhalt der in den Proben enthaltenen Zugänge Nachtschatten Abschnitt Petota Pangenom basierend auf PAV.
3. Gruppierung der Zugänge basierend auf dem Geninhalt
Phylogenetische Analysen und PCA identifizieren verschiedene Kladen und Untergruppen, die Variationen im Geninhalt widerspiegeln.
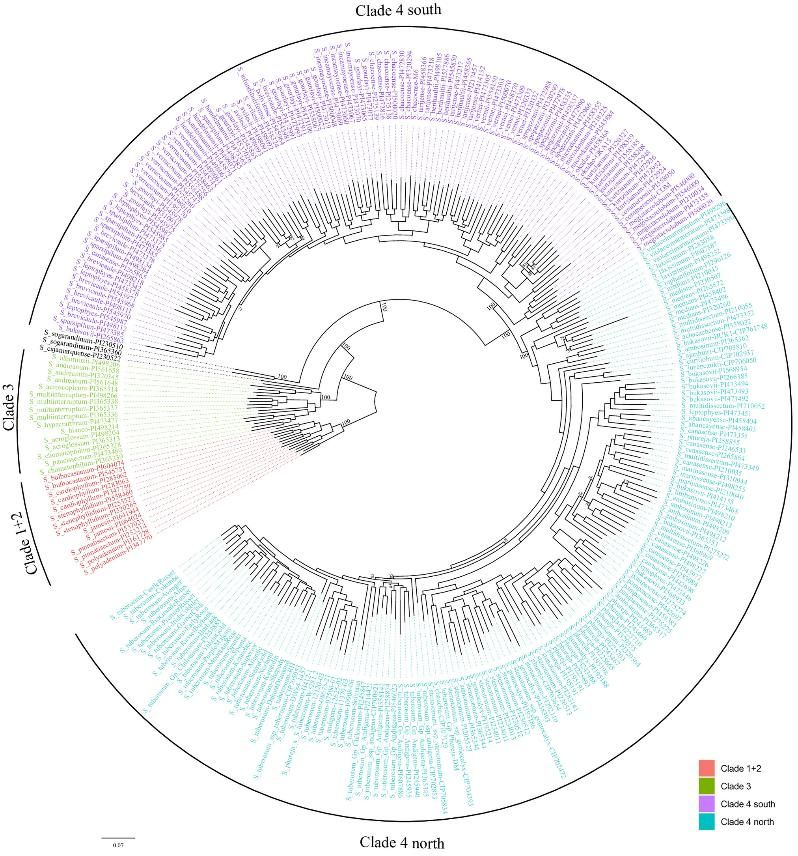 Abb. 3. Ein phylogenetischer Baum mit maximaler Wahrscheinlichkeit, der unter Verwendung von PAV-Daten für die Nachtschatten Abschnitt Petota Pangenom.
Abb. 3. Ein phylogenetischer Baum mit maximaler Wahrscheinlichkeit, der unter Verwendung von PAV-Daten für die Nachtschatten Abschnitt Petota Pangenom.
4. Variation im Geninhalt unterscheidet Kladen und Untergruppen
Die Unterschiede im Geninhalt sind zwischen den Kladen bemerkenswert und heben evolutionäre und Anpassungsmerkmale hervor.
5. TE-Inhalte im Pangenom
Das Pangenom enthält 75,5 % transposable Elemente, mit signifikanten kladenspezifischen Variationen und Unterschieden im TE-Inhalt.
Fazit
Die zukünftige Überlebensfähigkeit von Kartoffelpflanzen angesichts des Klimawandels hängt von der Erhaltung der Biodiversität und deren Integration in neue Sorten ab. Ein Pangenom von 296 Zugängen aus 60 Nachtschatten Abschnitt Petota Arten zeigen, dass PAV, insbesondere die Variation von TEs, eine Rolle bei der Artbildung spielt und einen erhöhten TE-Gehalt in in vitro propagierten Materialien aufweist, ähnlich der natürlichen stressinduzierten TE-Aktivität.
Referenz
- Bozan I, Achakkagari SR, Anglin NL, et al. Pangenom-Analysen zeigen den Einfluss von transponierbaren Elementen und Ploidie auf die Evolution von Kartoffelarten. Sitzungsberichte der Nationalen Akademie der Wissenschaften2023, 120(31):e2211117120.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Sammlung genetischer Daten in ethnisch basierten Studien bei Aymaras, Quechuas und Mestizen: die Herausforderungen der Genetik von Alzheimer in der peruanischen Bevölkerung (GAPP) Studie
Zeitschrift: Alzheimer & Demenz
Jahr: 2022
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibischer hispanischer Ethnie
Journal: JAMA Netzwerk Open
Jahr: 2023
Erhöhte Produktion pathogener, luftgetragener Pilzsporen bei der Exposition einer Bodenmykobiota gegenüber chlorierten aromatischen Kohlenwasserstoffschadstoffen
Journal: Mikrobiologie Spektrum
Jahr: 2023
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Journal: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


