
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben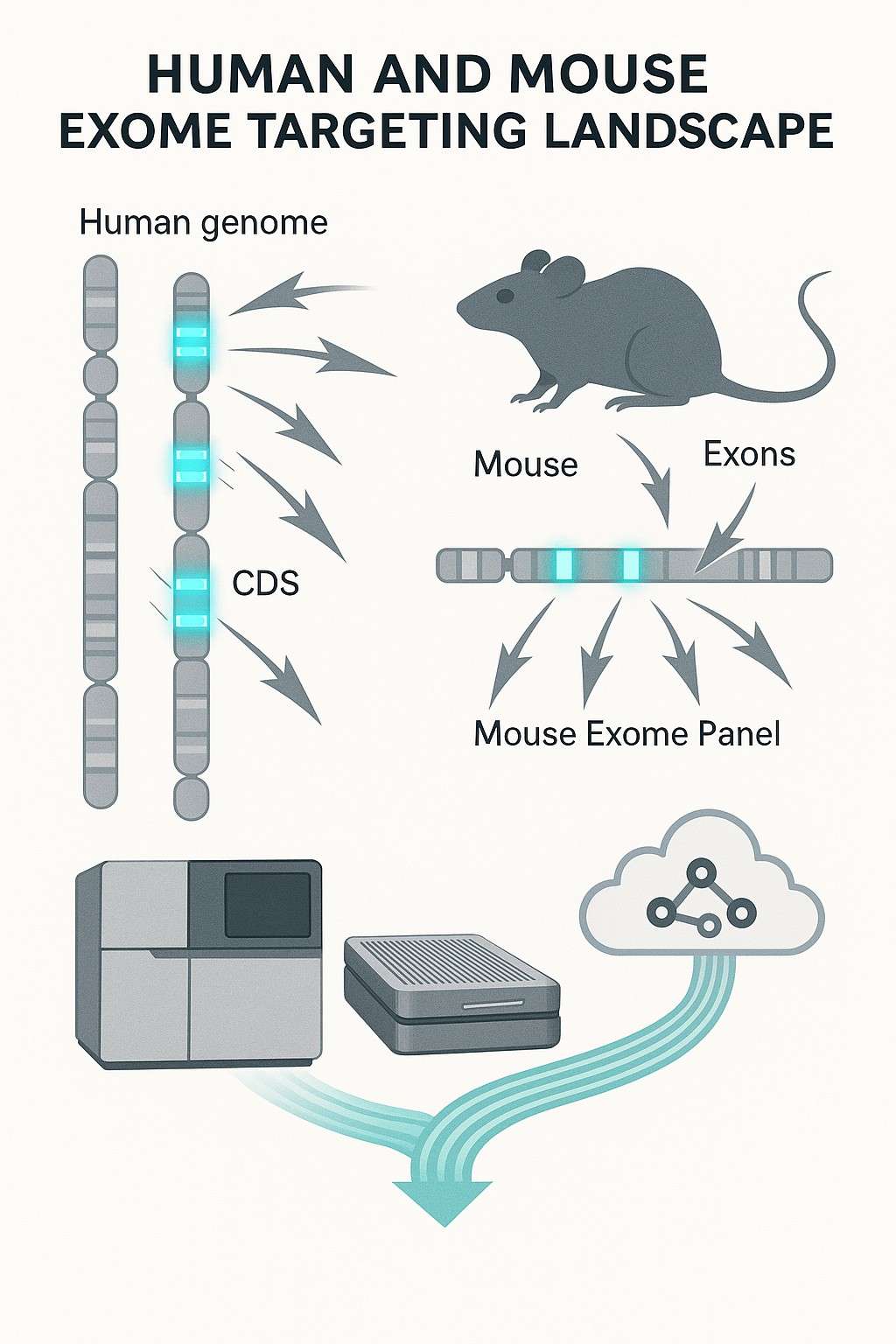
Warum die gesamte Exomsequenzierung von Mensch und Maus?
Whole-Exom-Sequenzierung (WES) konzentriert sich auf die kodierenden Regionen (Exons) des Genoms, die nur 1–2 % des menschlichen oder Mausgenoms ausmachen, aber über 85 % der bekannten krankheitsbezogenen Varianten enthalten. Durch die selektive Sequenzierung dieser wertvollen Regionen bietet WES eine kosteneffektive Alternative zu Whole-Genome-Sequenzierung (WGS) ohne die Entdeckungskraft für die meisten funktionalen Mutationen zu opfern.
Egal, ob Sie seltene Erbkrankheiten, Tumormutationen untersuchen oder Krankheitsmodelle in Mäusen entwickeln, Exom-Sequenzierung bietet tiefgehende Einblicke in protein-codierende Veränderungen, die Phänotyp und Pathologie antreiben.
Wesentliche Vorteile der Exom-Sequenzierung:
- Effizient und hochauflösend: Bestimmt Einzel-Nukleotid-Varianten (SNVs), Insertionen/Löschungen (InDels) und Kopienzahlvariationen (CNVs) in allen protein-codierenden Regionen.
- Kostengünstige Alternative zu WGS: Liefert umsetzbare Daten zu einem Bruchteil der Kosten und des Datenvolumens.
- Ideal für die Forschung zu menschlichen Krankheiten und Mausmodellen: Ermöglicht vergleichende Genomik, Mutationsscreening und translationale Erkenntnisse über Arten hinweg.
- Flexibel für Forschungsbedürfnisse: Unterstützt die Entdeckung von Keimbahn- und somatischen Varianten, Bevölkerungsstudien und gezielte funktionale Analysen.
- Erweiterte Variantenannotation: Nutzt umfassende Datenbanken wie RefSeq, ClinVar, CCDS und GENCODE.
Bei CD Genomics bieten wir sowohl an Human/Maus Whole Exome Sequenzierung und Pflanzen-/Tier-Whole-Exom-Sequenzierung, mit anpassbaren Tiefen-, Plattform- und Analyseoptionen, die auf Ihre spezifischen Forschungsziele zugeschnitten sind.
Unsere Exom-Sequenzierungsdienste für Menschen und Mäuse
Humanes Exom-Sequenzierungsdienst
Unser Menschliche Whole-Exome-Sequenzierung basiert auf GRCh38 und erfasst bis zu 99,9 % der bekannten protein-codierenden Regionen in den RefSeq-, MANE-, CCDS- und ClinVar-Datenbanken. Wählen Sie zwischen Standard- oder krankheitsfokussierten Optionen. Humane Exom-Panels:
- Kernpanel: Optimiert für hohe exone Abdeckung und effizienten Datenertrag.
- Erbkrankheiten-Panel: Fügt ClinVar-pathogene Stellen, mtDNA und Hochdichte-CNV-Probenrückgrate hinzu.
- Krebs-Panel: Enthält über 600 tumorassoziierte Gene, Fusionsregionen, HLA-Loci und MSI-Marker.
Wir unterstützen flexible Sequenzierungstiefen (100X–200X) mit Optionen für FFPE-, Blut- oder Gewebeproben.
Maus Exom-Sequenzierungsdienst
Unser Maus Whole Exome Sequenzierung verwendet ein proprietäres Maus Exom-Panel Entwickelt aus dem mm39-Genom, das über 38 Mb an CDS-Regionen mit hoher Erfassungsuniformität abdeckt. Dies ermöglicht eine effiziente Sequenzierung mit geringem Input und exzellenter Tiefe (100X+) unter Verwendung von nur 8 Gb Daten.
- Nutzen Sie unsere Mausplattform für:
- Phänotyp-zu-Genotyp-Studien
- Transgene und Knockout-Mausvalidierung
- Präklinische Krankheitsmodellanalyse
Human- und Maus-Exom-Sequenzierungs-Panels bei CD Genomics
| Art | Dienstname | Zielregionen | Empfohlene Datengröße | Notizen |
|---|---|---|---|---|
| Mensch | Humanes Exom-Sequencing – Kernpanel | ~34,4 Mb kodierende Sequenzen (CDS, GRCh38) | ≥8 Gb @100X | Optimiert für Abdeckung und Effizienz |
| Mensch | Humanes Exom-Sequencing – Erbliches Panel | CDS + pathogene Stellen (ClinVar, mtDNA, CNV-Regionen) | ≥11 Gb @100X | Verbesserte SNV/InDel/CNV-Erkennung |
| Mensch | Humanes Exom-Sequencing – Tumorpanel | CDS + 641 Krebsgenen, Fusions, MSI, HLA-Loci | ≥20 Gb @200X | Unterstützt TMB, MSI, Fusionsdetektion |
| Maus | Maus Exom-Sequenzierung – Standardpanel | ~38 Mb kodierende Regionen (basierend auf mm39) | ≥8 Gb @100X | Geeignet für die Phänotyp-Genotyp-Kartierung |
Technologieplattformen für Exom-Sequenzierung
Bei CD Genomics bieten wir umfassende Exom-Sequenzierung sowohl mit Short-Read- als auch mit Long-Read-Plattformen an, um unterschiedlichen Projektanforderungen in der Krankheitsforschung, der Arzneimittelentdeckung und der funktionellen Genomik gerecht zu werden.
Unterstützte Plattformen
- Illumina NovaSeq & NextSeq
Industrie-Standard für Kurzlesesequenzierung für Anwendungen mit hoher Tiefe und hohem Durchsatz.
Ideal zum Nachweis von Einzel-Nukleotid-Varianten (SNVs) und kleinen Insertionen/Deletion (InDels) in kodierenden Regionen. - MGI DNBSEQ
Eine kostengünstige Alternative zu Illumina, die vergleichbare Genauigkeit und Abdeckungsuniformität bietet.
Geeignet für Maus-Exomprojekte, die Skalierbarkeit und schnelle Bearbeitung erfordern. - Nanopore PromethION
Langzeit-Sequenzierungsplattform, die in der Lage ist, erweiterte Exons, Fusionsereignisse und komplexe strukturelle Varianten zu erfassen.
Nützlich zur Charakterisierung von Spleißisoformen, Genfusionen und schwer zu kartierenden GC-reichen Exons. - PacBio Revio / Sequel IIe (HiFi-Lesungen)
Ermöglicht hochpräzise Langreads (HiFi) für exakte Exon-Phasierung, CNV-Erkennung und de novo Assemblierung in gezielten Regionen.
Besonders vorteilhaft beim Erfassen von Volltexttranskripten und beim Lösen von Wiederholungen in menschlichen und Mausmodellen.
Lese-Längen und Abdeckung
| Plattform | Typische Lesezeit | Empfohlene Tiefe | Anwendungsnotizen |
|---|---|---|---|
| NovaSeq / DNBSEQ | PE150 | 100–200X | Standard-Exom-Variantenentdeckung |
| PromethION | 5–20 kB | 20–40X | Strukturelle Varianten, Phasierung, seltene Isoformen |
| PacBio HiFi | 10–25 kB | 30–50X | CNV-Auflösung, saubere Exon-Grenzen |
Exom-Capture-Strategien
- Hybridisierungsbasierte Erfassung
Optimierte Sonden-Sets, die entwickelt wurden, um kodierende Sequenzen aus den Genomen von Mensch und Maus anzureichern.
Benutzerdefinierte Panel-Optionen sind auf Anfrage für fokussierte Krankheitsgen-Sets oder orthologe Regionen verfügbar. - Amplicon-freie Arbeitsabläufe
Für degradierte oder FFPE-Proben verwenden wir enzymatische Fragmentierung und Protokolle mit niedrigem Input, um eine robuste Leistung sicherzustellen.
Arbeitsablauf
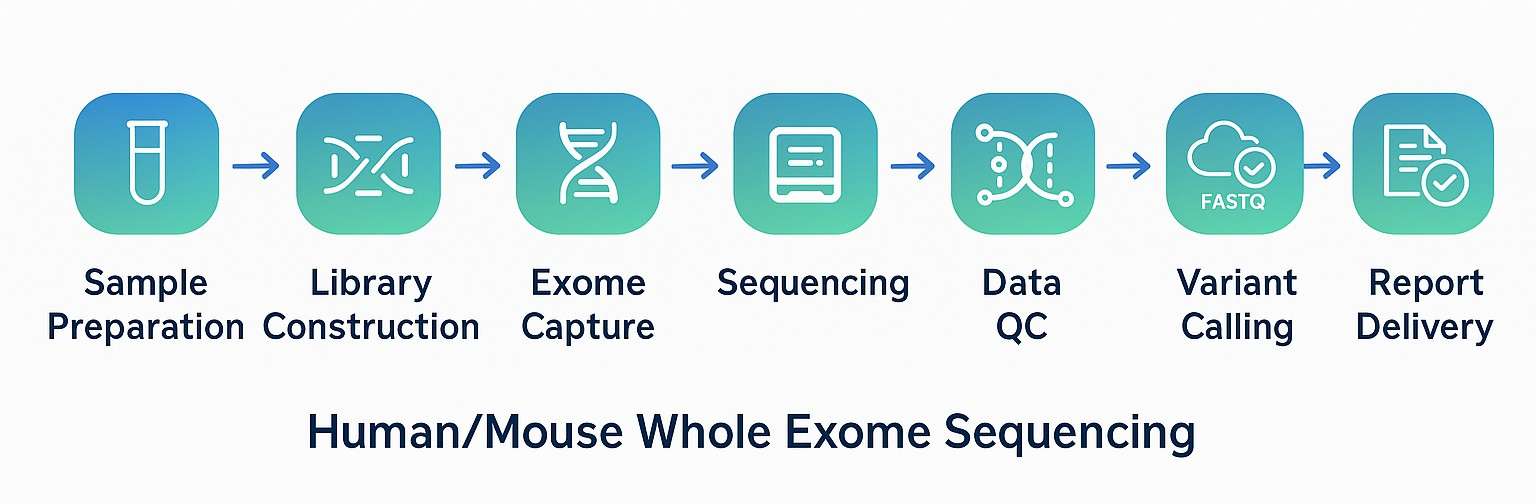
Datenanalyse
Unsere Bioinformatik-Pipeline liefert hochgradig zuverlässige Variantenaufrufe und umfassende Interpretationen für sowohl menschliche als auch Maus-Exome. Jedes Datenset wird mit Goldstandard-Algorithmen analysiert, um eine zuverlässige Erkennung klinisch und funktionell relevanter Varianten zu gewährleisten.
Was in unserer Exomdatenanalyse enthalten ist:
- Qualitätskontrolle von Roh-Sequenzierungsdaten (Phred-Score, Adaptertrimmen, Kontaminationsfilterung)
- Ausrichtung an das Referenzgenom (GRCh38 für Mensch, GRCm39 für Maus) mit BWA-MEM
- Duplikat-Markierung und Basisqualitäts-Rekalibrierung mit den GATK Best Practices
- Variantenerkennung (SNVs und kleine InDels) mit GATK HaplotypeCaller
- Funktionale Annotation mit Ensembl VEP und ClinVar/OMIM/gnomAD
- Seltene Variantenfilterung basierend auf Allelfrequenzschwellenwerten
- Pathogenitätsvorhersage mit Tools wie SIFT, PolyPhen-2, MutationTaster
- CNV-Analyse (für hochgradige Proben) mit CNVkit oder XHMM
- Tumorproben (optional): MSI/TMB-Analyse und somatische Mutationsbestimmung
Technischer Prozess
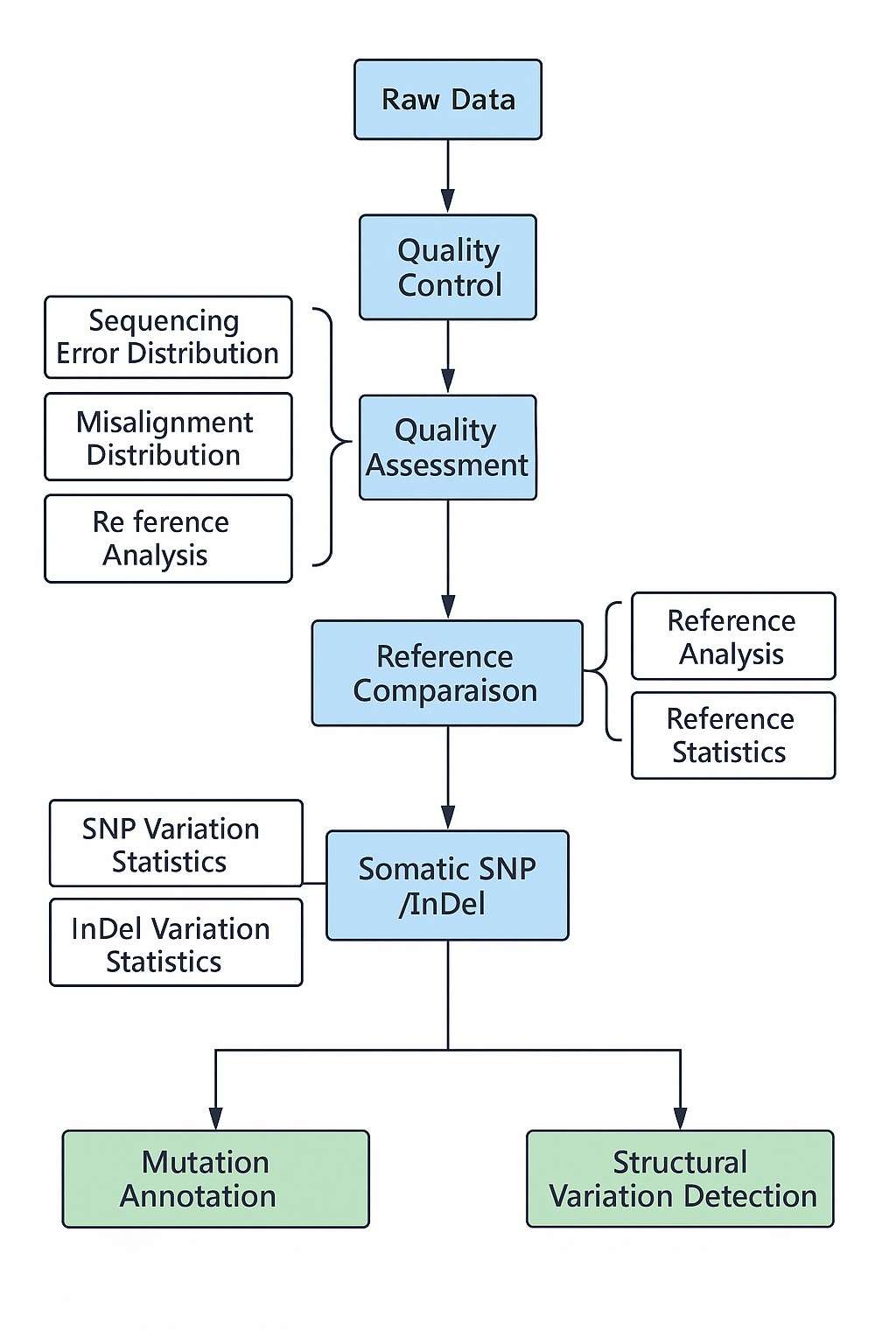
Fortgeschrittener technischer Prozess
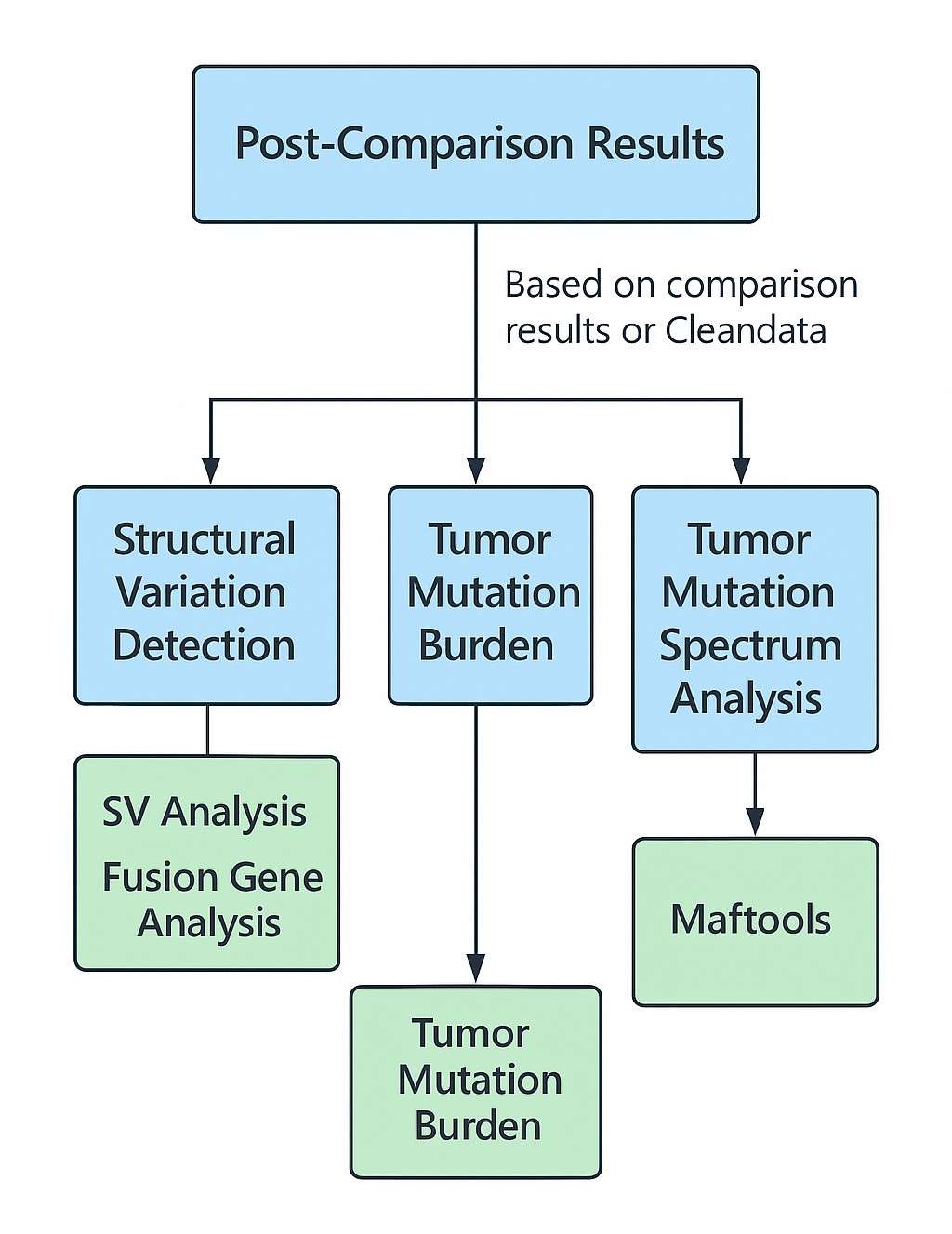
Vorteile unserer Exom-Sequenzierungsdienste
Unsere Exom-Sequenzierungsdienste sind auf Präzision, Flexibilität und interspezifische Kompatibilität optimiert. Egal, ob Sie seltene Krankheiten, Krebs oder funktionelle Genomik in Mausmodellen untersuchen, wir liefern hochwertige Daten – schnell.
Duale Artenexpertise
Validierte Exom-Panels für Mensch und Maus, die >99% der kodierenden Regionen abdecken (RefSeq, MANE, CCDS).
Flexible Plattformen
Illumina, MGI für kurze Reads; Nanopore, PacBio für lange Reads und komplexe Regionen.
Anpassbare Ziele
Fügen Sie HLA-, CNV-Scaffolds, das mitochondriale Genom oder krankheitsspezifische Regionen hinzu.
Hohe Uniformität und Empfindlichkeit
Ausgezeichnete Exon-Ebene-Abdeckung mit geringem Ausfall und hoher Erfassungs-effizienz.
Niedriger Input & FFPE-kompatibel
Optimiert für herausfordernde Proben mit nur 50 ng Eingangs-DNA.
Vollständige Bioinformatik-Pipeline
Beinhaltet SNV-, InDel-, CNV-Erkennung sowie optionale MSI/TMB-Analyse.
Beispielanforderungen
| Anwendung | Probenart | Empfohlene Menge | Mindestmenge | Mindestkonzentration |
|---|---|---|---|---|
| Human/Maus Whole Exome Sequenzierung | Genomische DNA | ≥ 500 ng | 100 ng | 10 ng/μL |
| PCR-freie Exom-Sequenzierung | Genomische DNA | ≥ 1 µg | 500 ng | 20 ng/μL |
| FFPE Exom (degradierte DNA) | FFPE-DNA | ≥ 500 ng | - | Fragment > 1000 bp |
Hinweis: Die Konzentrationen sollten durch Fluorometrie (Qubit, PicoGreen) bestimmt werden. Wenn Spektrophotometrie (z. B. NanoDrop) verwendet wird, verdoppeln Sie die Konzentrationswerte.
Wir akzeptieren eine Vielzahl von Probenarten und bieten auf Anfrage Extraktionsdienste an.
| Probenart | Menge | Versandbedingungen |
|---|---|---|
| Zellen | 1×10⁶ | Trockeneis |
| Frisch gefrorenes Gewebe | 10 mg | Trockeneis |
| FFPE-Folien | ≥4 Folien (≥150 mm²) | Raumtemperatur / Blauer Eis |
| Blut (EDTA-Röhrchen) | 2–4 ml | Blaues Eis / Trockeneis |
| Plasma / Serum | 10 ml | Trockeneis |
| Speichel | 1 ml | Trockeneis / Blausee |
| Stuhl / Erde | 100 mg | Trockeneis / Raumtemperatur |
| Wattestäbchen | 2 Röhrchen / Probe | Raumtemperatur |
| Wasser | 50 ml | Raumtemperatur |
Sind Sie sich nicht sicher, ob Ihre Probe qualifiziert? Kontaktieren Sie uns für eine kostenlose Beratung und Unterstützung bei der Entnahme.
Was Sie erhalten werden
- Die Liefergegenstände sind auf den Umfang Ihres Projekts zugeschnitten:
- Rohdaten-Dateien (FASTQ)
- Ausrichtungsdateien (BAM) und Variationsdateien (VCF)
- Statistische und Annotationsberichte (PDF + Excel)
- Grafische Analyseergebnisse
- Projektdokumentation und Nutzungshinweise
FAQs zur gesamten Exom-Sequenzierung von Mensch und Maus
Q1: Warum sollte man sich für die gesamte Exomsequenzierung anstelle der gesamten Genomsequenzierung entscheiden?
A: Die gesamte Exomsequenzierung (WES) konzentriert sich auf die etwa 1–2 % des Genoms, die für Proteine kodieren, erfasst jedoch etwa 85 % der krankheitsverursachenden Mutationen. Es ist eine kosteneffektive Lösung, wenn Ihr Forschungsschwerpunkt funktionale Variationen auf Genebene umfasst.
Q2: Was ist der Unterschied zwischen Ihren Dienstleistungen für die Sequenzierung des menschlichen Exoms und des Maus-Exoms?
A: Beide Dienstleistungen umfassen die probe-basierte Erfassung von protein-codierenden Regionen (CDS), Hochdurchsatz-Sequenzierung und Varianteninterpretation. Die Humanes Exom-Panel ist auf GRCh38 ausgerichtet und enthält ClinVar-Anmerkungen, während die Maus Exom-Panel Ziele GRCm39 und unterstützt Studien zu Krankheitsmodellen.
Q3: Welche Deckungstiefe empfehlen Sie für die Exom-Sequenzierung von Menschen oder Mäusen?
A: Wir empfehlen 100X–150X für Keimbahnstudien und ≥200X für Tumor-/FFPE-Proben. Für Maus Whole-Exom-SequenzierungEine tiefere Abdeckung kann für mosaik- oder chimäre Modelle erforderlich sein.
Q4: Unterstützen Sie Long-Read-Exom-Sequenzierung mit Nanopore oder PacBio SMRT?
A: Ja. Für ausgewählte Anwendungen, die die Erkennung von strukturellen Varianten oder hochrepetitiven Regionen erfordern, bieten wir an Long-Read-Exom-Capture Verwendung von Nanopore- oder SMRT-Plattformen. Bitte kontaktieren Sie uns für maßgeschneiderte Protokolle.
Q5: Wie stellen Sie die Variantenaufrichtigkeit in FFPE- oder Low-Input-Proben sicher?
A: Wir verwenden optimierte Bibliotheksvorbereitungs-Kits mit UMI (einzigartigen molekularen Identifikatoren), FFPE-geeigneten Capture-Panels und maßgeschneiderten bioinformatischen Pipelines, um die Sensitivität zu verbessern und Artefakte zu reduzieren.
Q6: Kann ich vorangereicherte Exom-Bibliotheken nur zur Sequenzierung einreichen?
A: Ja. Wir akzeptieren sowohl rohes genomisches DNA als auch vorab erstellte Bibliotheken. Bitte kontaktieren Sie uns bezüglich der Anforderungen an die Qualitätskontrolle der Bibliotheken.
Q7: Welche Ergebnisse werde ich nach der Analyse erhalten?
A: Sie erhalten Rohdaten (FASTQ), ausgerichtete Dateien (BAM), Variantenaufrufe (VCF), annotierte Variantenlisten (Excel) sowie optionale CNV- oder Pfadberichte.
Q8: Wie wähle ich zwischen der gesamten Exom- und der gesamten Genomsequenzierung?
A: Wählen Whole-Genome-Sequenzierung wenn Ihre Studie die Analyse von nicht-kodierenden Varianten, strukturellen Variationen oder intergenen regulatorischen Elementen erfordert. Für die Entdeckung von Mutationen auf Genebene, menschliche ganze Exom-Sequenzierung bietet höhere Tiefe zu geringeren Kosten.
Fallstudien zur gesamten Exomsequenzierung von Mensch und Maus
Kundenveröffentlichungshighlight
Fallstudie: Integriertes Exom- und optisches Mapping bei klarzelligem Nierenzellkarzinom
Titel: Optische Genom- und Epigenomkartierung von klarzelligem Nierenzellkarzinom
Tagebuch: NAR Krebs
Veröffentlicht: 4. März 2025
DOI: 10.1093/narcan/zcaf008
Hintergrund
Das klarzellige Nierenzellkarzinom (ccRCC) ist die häufigste Form von Nierenkrebs, die oft durch komplexe genomische Ereignisse wie den Verlust von Chromosom 3p und die Inaktivierung des VHL-Gens bedingt ist. Allerdings spielen auch epigenetische Faktoren – einschließlich DNA-Methylierung und Hydroxymethylierung – eine entscheidende Rolle bei der Tumorprogression. Die Erfassung beider Variationsarten in einem einzigen Workflow ist mit traditionellen Sequenzierungsmethoden herausfordernd.
Projektziele
- Hybride Charakterisierung: Kombinieren Sie die Kurzlese-Exomsequenzierung mit der optischen Genomabbildung, um sowohl kodierende Mutationen als auch großflächige strukturelle Variationen (SVs) zu erkennen.
- Epigenetisches Profiling: Messung von Veränderungen der Einzelmolekül-Methylierung (5mC) und Hydroxymethylierung (5hmC) im Tumor- im Vergleich zu normalem Gewebe.
- Klinische Einsicht: Verknüpfen Sie genetische und epigenetische Veränderungen mit funktionalen Genveränderungen für die Pathogenese des ccRCC.
CD Genomics Dienstleistungen
1. Exom-Sequenzierung
- Proben: Tumor- und passende benachbarte normale Gewebe
- Plattform: Humane Whole Exome Sequenzierung – Kernpanel (Illumina PE150, ~100 × Abdeckung)
- Analyse: GATK-basierte Variantenbestimmung (SNV/InDel) mit fokussierter Annotation von VHL, PBRM1, SETD2 und anderen epigenetischen Treibergenen.
2. Optische Genom-/Epigenom-Kartierung
(Diese Komponente wurde vom Forschungsteam durchgeführt)
- Erkennung struktureller Varianten mit Einzelmolekülauflösung
- Einzelmolekül-Methylierungsprofilierung mit markierten DNA-Molekülen
3. Integrierte Analyse
- Kreuzvalidierung von exom-detected Varianten mit Kopienzahl- und strukturellen Befunden aus der optischen Abbildung
- Vergleichende epigenomische Profilierung der 5mC- und 5hmC-Spiegel in wichtigen Tumorsuppressoren
Wichtigste Erkenntnisse
Koinzidente genetische und epigenetische Ereignisse
- Identifizierung einer 3p-Deletion, die VHL und PBRM1 umfasst, bestätigt durch optische Kartierung.
- Die Exom-Sequenzierung ergab nicht-synonyme Mutationen in SETD2 und BAP1, die in beiden Tumorstadien konsistent sind.
Epigenetische Dysregulation
- Globale Reduktion von 5hmC im Tumor im Vergleich zu normalem Gewebe.
- Hypo-/hypermethylierungsmuster in den Promotor-/Enhancerregionen von VHL, PRCC und PBRM1 korrelierten mit einer reduzierten Expression.
Funktionale genomische Einblicke
- Kombinierte Daten zeigten gestörte Stoffwechsel- und Hypoxiewege, die zur Progression von ccRCC beitragen.
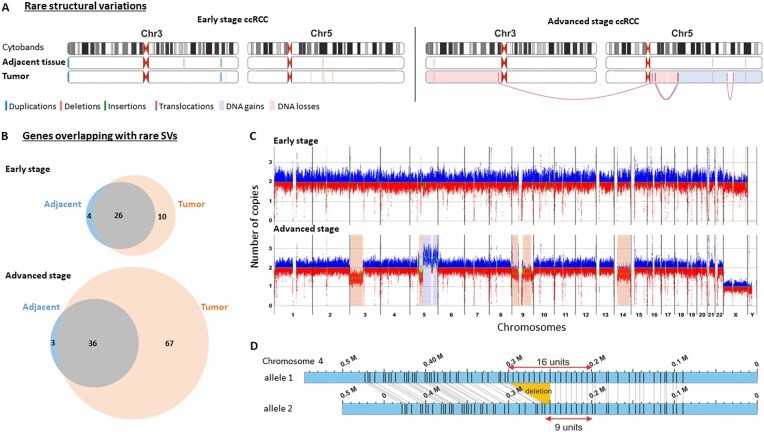 Dieses Panel zeigt die strukturelle Variantenkarte des Chromosoms 3p (Tumor vs. Kontrolle) und zeigt deutlich die 3p-Deletion, die durch optische Genomkartierung nachgewiesen wurde.
Dieses Panel zeigt die strukturelle Variantenkarte des Chromosoms 3p (Tumor vs. Kontrolle) und zeigt deutlich die 3p-Deletion, die durch optische Genomkartierung nachgewiesen wurde.
Implikationen
- Umfassende Profilierung: Die Kombination von Exom-Sequenzierung mit optischem Genom-Mapping ermöglicht eine vollständige Abdeckung – von Punktmutationen bis hin zu großen strukturellen Veränderungen.
- Erweiterte klinische Einblicke: Epigenetische Marker (5hmC/5mC) bieten zusätzliche Schichten der Tumorbiologie, die zuvor nur durch NGS-Ansätze verborgen waren.
- Strategischer Ansatz in der Krebsforschung: Diese Dual-Modality-Methode geht effektiv darauf ein, warum die gesamte Exomsequenzierung allein kritische Veränderungen bei Krebsarten wie ccRCC übersehen könnte.
Warum das für Sie wichtig ist
Dieser Fall unterstreicht die Kraft der Integration. Humanes Exom-Sequencing (für SNVs und InDels) mit langfristige strukturelle und epigenetische Kartierung um Krebsgenome vollständig zu entschlüsseln. Bei CD Genomics können wir Ihnen helfen, solche integrativen Strategien umzusetzen – indem wir sowohl Exom-Panels und Longread-Plattformen wie Nanopore oder PacBio, ergänzt durch Partnerschaften im Bereich der optischen Genomkartierung.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Eine de novo-Assemblierung von genomischen Datensatzsequenzen der Zuckerrübenfliegenlarve. Tetanops myopaeformis, TmSBRM_v1.0
Tagebuch: Daten im Überblick
Jahr: 2024
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neugeborenen Schweineherzen regulieren
Tagebuch: JCI Insight
Jahr: 2024
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Keime. Pseudomonas aeruginosa und die Empfindlichkeit gegenüber Carbapenem-Antibiotika erhöhen
Tagebuch: Viren
Jahr: 2024
Genomsequenz, Antibiotikaresistenzgene und Plasmide in einer monophasen Variante von Salmonella typhimurium vom Einzelhandel isoliertes Schweinefleisch
Journal: Ankündigungen zu Mikrobiologie-Ressourcen
Jahr: 2024
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


