
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben

Was ist das bakterielle Gesamtegenom? von Neuem Sequenzierung
Bakterielles Gesamtegenom von Neuem Sequenzierung ist ein referenzfreier Ansatz, der eine vollständige Rekonstruktion bakterieller Genome – einschließlich sowohl Chromosomen als auch Plasmide – direkt aus Stichprobendaten ermöglicht.
Diese Technik liefert eine vollständige, hochauflösende Genomkarte, die sie ideal für das Studium unbekannter Stämme, die Identifizierung von Genfunktionen und die Analyse mikrobieller Evolution macht. Sie ist besonders nützlich, wenn kein zuverlässiges Referenzgenom vorhanden ist oder wenn es sich um genetisch komplexe Arten handelt.
Wie es funktioniert: Vom Sample zum vollständigen Genom
Der von Neuem Der Sequenzierungsprozess integriert Langzeit-Sequenzierung, hochgenaue Korrektur von Kurzlesungen und robuste Zusammenstellungstools, um Präzision in jedem Schritt zu gewährleisten:
- Langzeit-Sequenzierung
Bibliotheksvorbereitung
Plattformen wie PacBio HiFi oder Oxford Nanopore erzeugen kontinuierliche Reads von 10–25 kb oder länger. Diese langen Reads umfassen sich wiederholende und strukturell komplexe Regionen, die mit kurzen Reads allein schwer zu entschlüsseln sind. - Referenzfreie Genomassemblierung
Montagewerkzeuge wie Hifiasm und Canu fügen diese langen Reads zu vollständigen Genomen zusammen – ganz von Grund auf, ohne auf vorhandene Referenzsequenzen angewiesen zu sein. - Kurzlese-Politur
Hochgenaue Daten aus der Illumina-Sequenzierung werden hinzugefügt, um kleinere Fehler zu korrigieren, wodurch die Zuverlässigkeit und die Basisgenauigkeit der Assemblierung verbessert werden. - Mehrstufige Genomannotation
Die abschließenden Qualitätskontroll- und funktionalen Annotationspipelines stellen sicher, dass Ihre Daten nicht nur vollständig, sondern auch biologisch sinnvoll sind – bereit für die nachgelagerte Analyse.
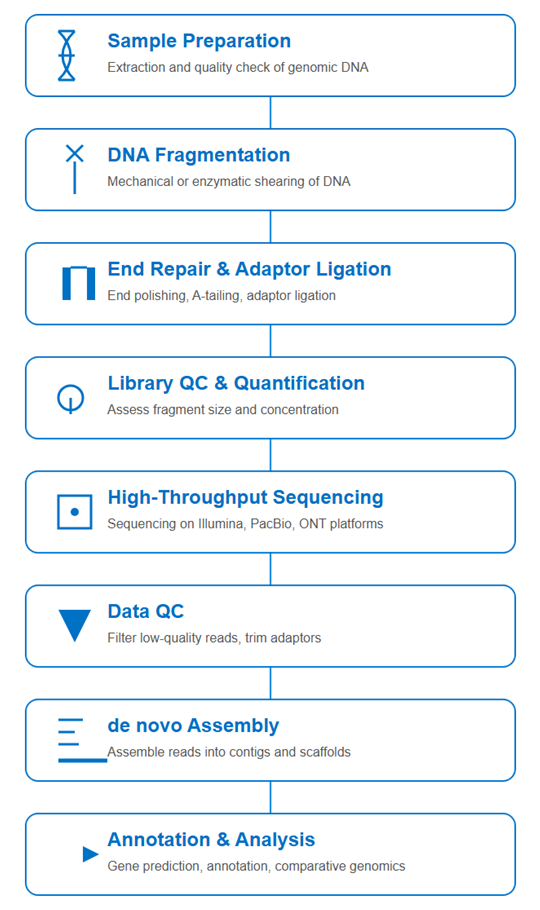 Ganzes Genom von Neuem Sequenzierungsprozess
Ganzes Genom von Neuem Sequenzierungsprozess
Warum das bakterielle Gesamtgenom von neuem Sequenzierung ist entscheidend für Ihre Forschung.

Speziell für bakterielle Proben entwickelt, die über kein Referenzgenom oder nur begrenzte Referenzdaten verfügen, von Neuem Die Ganzgenomsequenzierung ermöglicht eine genaue und vollständige Genomassemblierung. Dieser Ansatz offenbart komplexe genetische Variationen und repetitive Regionen, was die Qualität und Genauigkeit der Assemblierung erheblich verbessert.
- Genaues, vollständiges Genom-Assembly
Stellen Sie Chromosomen und Plasmide mit hoher Kontinuität zusammen – ohne auf Referenzsequenzen angewiesen zu sein. - Umfassende Genomanalyse
Erkennen Sie strukturelle Variationen, funktionale Gene, Marker für antimikrobielle Resistenzen und repetitive Elemente für ein vollständiges genetisches Profil. - Integration fortgeschrittener Sequenzierungstechnologien
Kombinieren Sie Langzeit-Sequenzierung mit hochdurchsatzfähigen Kurzlesungen, um Datengenauigkeit und -tiefe sicherzustellen. - Vielseitig für verschiedene Stämme
Geeignet für neuartige, komplexe oder schwer zu sequenzierende Bakterienstämme, die zuverlässige Zusammenbau-Ergebnisse garantieren.
Unser gesamtes Genom neu Sequenzierungsportfolio: Maßgeschneidert für Bakterien, Pilze und mehr
CD Genomics bietet artspezifische von neuem Genomsequenzierungsdienste, um vielfältige Forschungsbedürfnisse zu erfüllen, ohne die Einschränkungen eines Referenzgenoms.

Bakterielles Gesamtgenom von Neuem Sequenzierung
Referenzfreie Assemblierung | Vollständige Genomrekonstruktion | Entdeckung struktureller Variationen
Bakterielle WGS-Details anzeigen ↓
Pilz-Ganzgenom von Neuem Sequenzierung
Hochkontiguitätsassemblierung | Wiederholungsreiche Genomauflösung | Funktionsannotation bereit
Erforschen Sie den Fungal WGS-Service →
De Novo Whole Genome Sequenzierungsdienst
Multi-Spezies-Unterstützung | Integration von Lang- und Kurzlesungen | Ideal für neue Arten
Erfahren Sie mehr über Mehrarten. von Neuem WGS → WGSOptimierter Workflow für das bakterielle Whole Genome von Neuem Sequenzierung: Von der Probe zu Erkenntnissen

≥10 µg hochqualitative DNA
OD260/280 = 1,8–2,0

PacBio / Nanopore / Illumina
Lange und kurze Einfügebibliotheken

von neuem Montagewerkzeuge
Mehrstufige Politur
Hybride Fehlerkorrektur

Genvorhersage und Annotation
Identifizierung von Resistenz-/Virulenzgenen
Funktionale und vergleichende Genomik

Qualitätskontrollkennzahlen
Visuelle Berichte und Datenzusammenfassung
Optimierte Sequenzierungsstrategien für das gesamte bakterielle Genom von neuem Versammlung
Höhepunkte des Bibliotheksbaus:
- Mehrere Insertgrößenbibliotheken, die für optimale Abdeckung ausgelegt sind, einschließlich kurzer Inserts (~350 bp) und langer Inserts (5–20 kb).
- PCR-freie Bibliotheksvorbereitung zur Minimierung von Amplifikationsbias.
- Strenge Qualitätskontrolle zur Sicherstellung einer gleichmäßigen Datenabdeckung.
Sequenzierungsplattformen:
- PacBio Sequel IIe / Revio: Produziert hochgenaue HiFi-Langreads (10–25 kb) mit >Q20 Genauigkeit, ideal für die kontinuierliche Assemblierung komplexer Genome.
- Oxford Nanopore PromethION: Bietet ultra-lange Reads, die Megabasisskala erreichen und die Assemblierung hochkomplexer Regionen verbessern.
- Illumina NovaSeq 6000: 150 bp Paar-End-Sequenzierung mit tiefem Coverage und >90% Basen in Q30-Qualität, perfekt für Fehlerkorrektur und präzises Polieren.
Empfohlene Sequenzierungstiefe:
- Standard: >100× Abdeckung mit PacBio HiFi-Lesungen.
- Zusätzlich: >50× Abdeckung mit Illumina-Kurzlesungen, um eine hochgenaue Genomkorrektur zu gewährleisten.
Datenqualitätsmetriken:
- HiFi-Lesegenauigkeit über 99,9 %.
- Illumina-Daten mit über 90 % der Basen in Q30-Qualität oder höher.
- Hohe Datenintegrität mit hervorragender Montagekontinuität.
Fortgeschrittene Bioinformatikanalyse: Bakterielle Genomdaten in umsetzbare Erkenntnisse umwandeln
Wir bieten professionelle, effiziente und umfassende Dienstleistungen an. Bioinformatikanalyse Dienstleistungen zur Ausschöpfung des vollen Potenzials Ihrer bakteriellen Genomdaten und zur Beschleunigung des Forschungsfortschritts.
Standardanalyse – Gewährleistung von Datenqualität und -genauigkeit
- Datenqualitätskontrolle und -bereinigung: Entfernen Sie niedrigqualitative und kontaminierte Sequenzen, um eine zuverlässige nachgelagerte Analyse zu gewährleisten.
- Genomassemblierung und Scaffoldierung: Verwenden Sie fortschrittliche Algorithmen, um kontinuierliche, vollständige bakterielle Genomsequenzen zu erzeugen.
- Sequenzkorrektur: Führen Sie mehrere Runden der Fehlerkorrektur durch, um Sequenzierungsfehler zu reduzieren und die Annotationsqualität zu verbessern.
- Genvorhersage: Genau identifizieren Sie protein-codierende Gene und nicht-codierende RNAs für ein umfassendes Verständnis der Genomfunktion.
- Funktionale Annotation: Integrieren Sie Datenbanken wie GO, KEGG und eggNOG, um biologische Rollen von Genen und Stoffwechselwege zu definieren.
- Wiederholte Sequenzen und CRISPR-Vorhersage: Erkennen Sie komplexe Genomstrukturen und bakterielle Abwehrsysteme für tiefgehende funktionale Einblicke.
Fortgeschrittene Tiefenanalyse – Gewinnung zusätzlicher biologischer Erkenntnisse
- Virus- und Phagenvorhersage: Identifizieren Sie potenzielle Prophage-Sequenzen innerhalb bakterieller Genome, um mikrobielle Interaktionen aufzudecken.
- Virulenzfaktoren und Resistenzgenanalyse: Präzise Erkennung von Virulenz- und Antibiotikaresistenzgenen zur Unterstützung von Erregerstudien und Resistenzüberwachung.
- Analyse der kohlenhydrataktiven Enzyme (CAZy): Untersuchen Sie Gene, die für metabolische Enzyme kodieren, um die Forschung zu industriellen Enzymen und dem Stoffwechsel zu unterstützen.
- Transmembranproteine und Vorhersage von Signalpeptiden: Vorhersage wichtiger Membranproteine und Signalpeptide zur Unterstützung der Entdeckung von Arzneimittelzielen und funktionalen Studien.
- Vergleichende Genomik: Führen Sie phylogenetische Bäume, Genfamilien-Clustering und Syntenie-Analysen durch, um die Evolution von Stämmen und funktionale Unterschiede zu untersuchen.
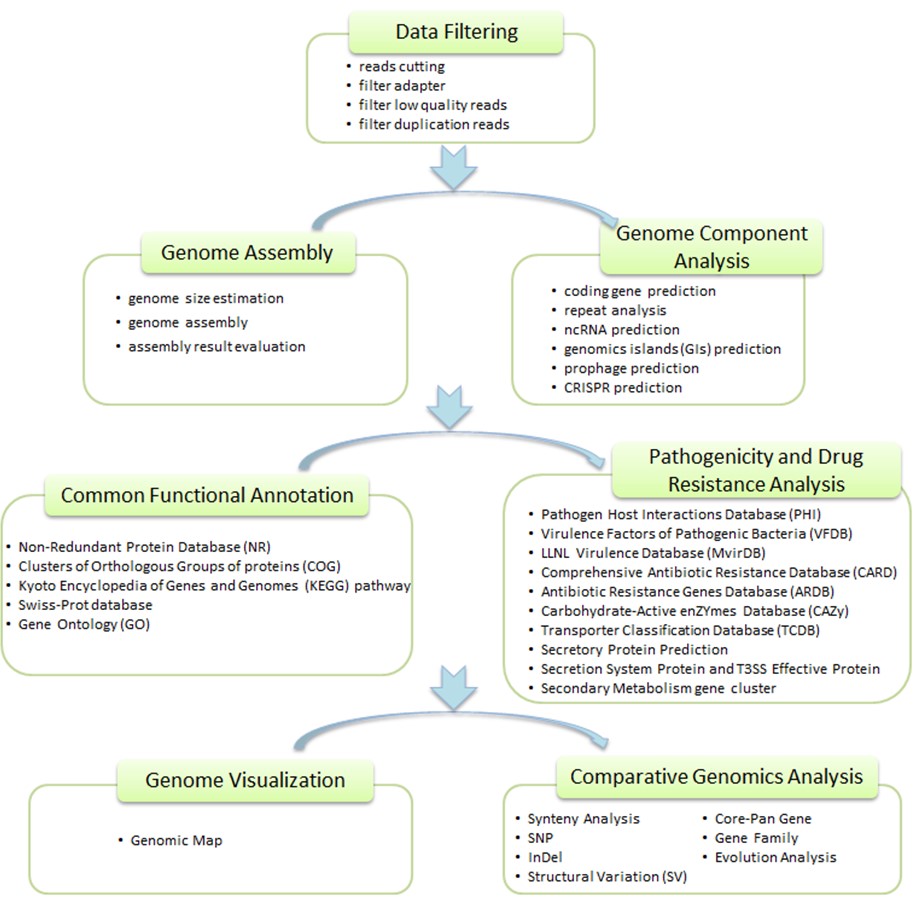
Beispielanforderungen für das bakterielle Gesamtegenom von Neuem Sequenzierung
| Probenart | Anforderungsbeschreibung |
|---|---|
| Gesamtmenge an DNA | ≥ 10 μg |
| DNA-Konzentration | ≥ 80 ng/µL |
| DNA-Reinheit | OD260/OD280-Verhältnis zwischen 1,8 und 2,0 |
| Integrität | Keine sichtbare Degradation oder RNA-Kontamination; durch Gelelektrophorese auf Unversehrtheit überprüft. |
Empfehlungen zur Einreichung von Proben:
- Verwenden Sie DNase-freie, niedrig-bindende Zentrifugentuben, wie z. B. 1,5 mL Eppendorf-Tuben, um Proben zu lagern.
- Für den kurzfristigen Transport die Proben mit Kühlakkus kühl halten; für längeren Transport Trockeneis verwenden.
- Kennzeichnen Sie jede Probe deutlich mit einer identifizierbaren Nummer.
Anwendungen des bakteriellen Gesamten Genoms von Neuem Sequenzierung
Unser bakterielles Gesamtgenom von Neuem Der Sequenzierungsdienst bietet einen umfassenden Überblick über bakterielle Genome und unterstützt eine Vielzahl von Forschungsbereichen:

- Mechanismen der Antibiotikaresistenz
Präzise Lokalisierung von Resistenzgeninseln wie β-Lactamasen, um das Verständnis von Arzneimittelresistenz zu fördern und öffentliche Gesundheitsinterventionen zu leiten. - Verfolgung der Virulenzentwicklung
Analysieren Sie den horizontalen Transfer von Virulenzfaktoren, um Studien zur Evolution von Pathogenen und zu Veränderungen der Virulenz zu unterstützen. - Industrielle Spannungsoptimierung
Identifizieren Sie Schlüsselgene der Stoffwechselwege, um die Fermentationseffizienz zu steigern und bakterielle Stämme zu verbessern. - Umweltanpassungsmechanismen
Enthüllen Sie Überlebensstrategien von Bakterien in extremen Umgebungen, die der ökologischen und umweltbezogenen Forschung zugutekommen. - Neue Artenidentifikation
Erstellen Sie vollständige genomische Profile, um die mikrobielle Taxonomie zu erleichtern und neuartige Arten zu entdecken.
Warum CD Genomics für bakterielle Gesamte Genom-Analyse wählen? von Neuem Sequenzierung?
CD Genomics bietet einen vertrauenswürdigen, umfassenden Service mit hochwertiger, schneller Bearbeitung und umfassender Analyse für bakterielle Vollgenomsequenzierung an. von Neuem Sequenzierung. Unser Fokus geht über die Sequenzierung hinaus und umfasst die Gewährleistung einer erstklassigen Datenqualität und biologisch sinnvoller Ergebnisse.
- Multi-Plattform integrierte Sequenzierungsstrategie
Kombinieren Sie die Stärken der PacBio HiFi-, Oxford Nanopore- und Illumina-Plattformen, um eine hohe Genauigkeit und vollständige Genomassemblierungen zu erreichen. - Angepasste Montage- und Korrektur-Pipelines
Passen Sie die Zusammenstellungsprotokolle basierend auf den Probenmerkmalen an, indem Sie mehrere Runden der Fehlerkorrektur mit Rohdaten der dritten Generation, Selbstanpassung und Politur der Daten der zweiten Generation anwenden, um hochgenaue Endsequenzen sicherzustellen. - Umfassende bioinformatische Analysen
Von der Verarbeitung roher Daten über funktionelle Annotation, Phylogenetik, Resistenzgene, Virulenzfaktoren bis hin zu Stoffwechselwegen unterstützen unsere Analysen vielfältige Forschungsziele. - Strenges Qualitätskontrollsystem
Befolgen Sie standardisierte Protokolle vom Erhalt der Proben über die Bibliotheksvorbereitung, Sequenzierung und Qualitätskontrolle der Assemblierung, um eine konsistente und zuverlässige Datenlieferung zu gewährleisten. - Klare, visuelle und umsetzbare Datenberichte
Stellen Sie leicht verständliche Diagramme und strukturierte Annotationsdateien bereit, die nachgelagerte Analysen und die Manuskriptvorbereitung erleichtern. - Engagierter technischer Support
Erhalten Sie fachkundige Unterstützung während Ihres Projekts, einschließlich experimenteller Gestaltung, Dateninterpretation und Fehlersuche, die Ihnen hilft, komplexe Analyseherausforderungen reibungslos zu meistern.

Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
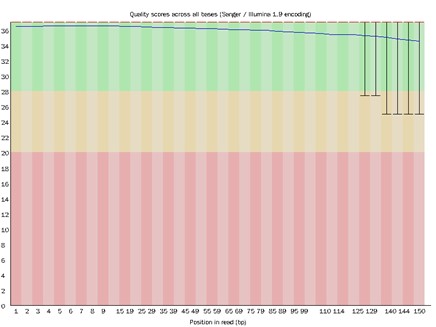
Verteilung der Basisqualität
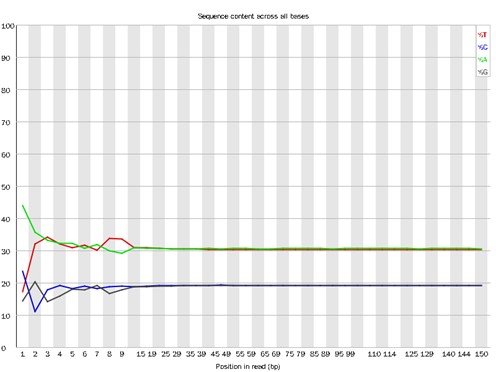
Verteilung des Basisinhalts
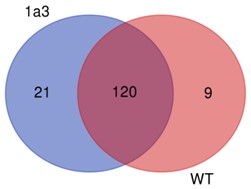
Geteilte SNP-Nummer zwischen Proben
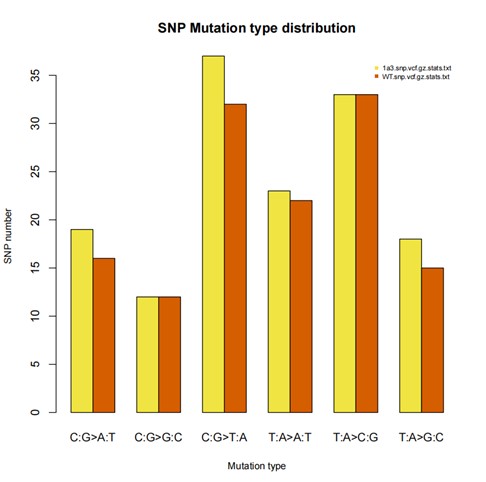
Verteilung der SNP-Mutationsarten
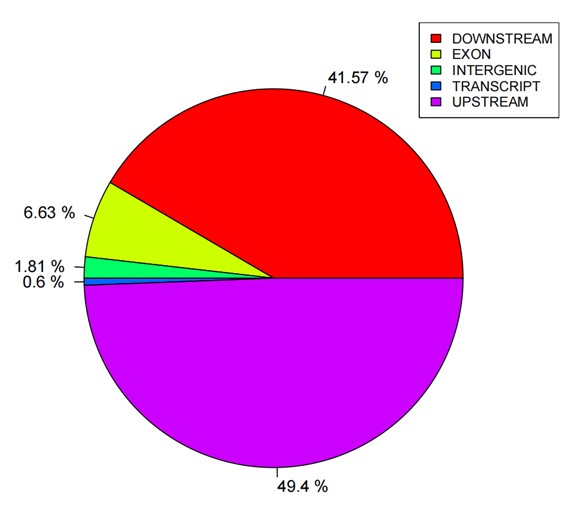
Statistik-Kreisdiagramm der SNP-Anmerkungen
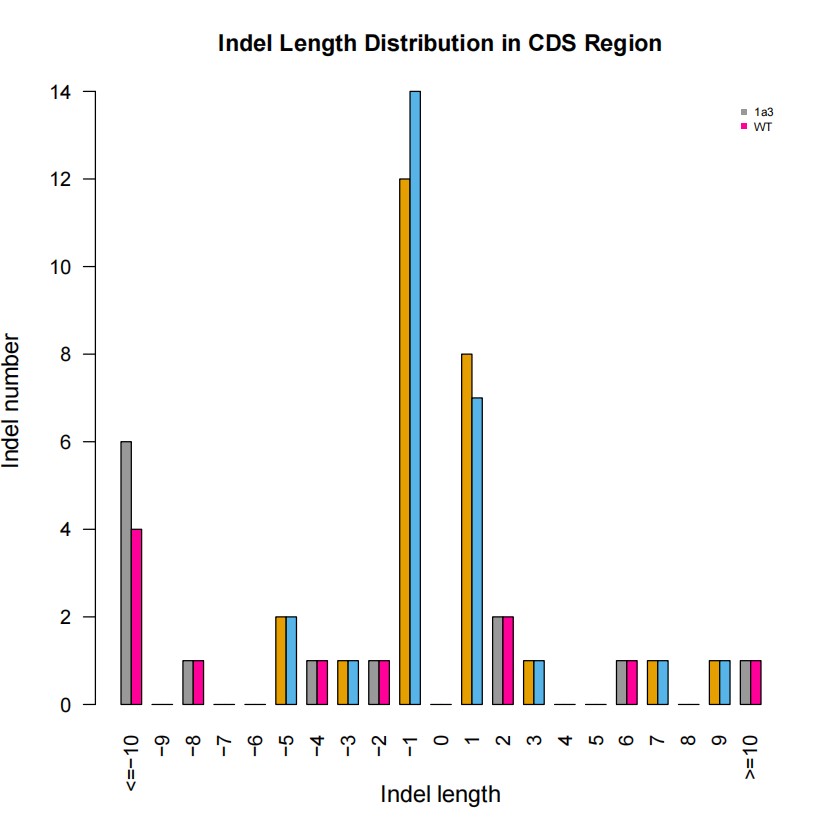
InDel-Längenverteilung
Bakterielle Gesamtegenom von Neuem Seq FAQs
1. Welche Indikatoren können verwendet werden, um die Assemblierung von bakteriellen Genomen zu bewerten?
Die gängigen Indikatoren für die Qualität der Genomassemblierung umfassen Scaffold N50, N%, die Anzahl der Scaffolds und die Gesamtzahl der Basenpaare.
2. Wie erreicht man einen Nullabstand?
Derzeit kann die vollständige Sequenzkarte von mehr als 90 % der bakteriellen Stämme durch die Kombination von Illumina HiSeq- und PacBio SMRT-Systemen erstellt werden. Das PacBio RS II-System kann eine vollständige Genomassemblierung sogar in Regionen mit hohem oder niedrigem GC-Gehalt sowie in repetitiven Sequenzen erreichen. Die vollständige Sequenzkarte der restlichen 10 % der bakteriellen Stämme kann mit Sanger-Sequenzierungsdaten erstellt werden. CD Genomics hat Hunderte von Fällen der bakteriellen Genomassemblierung ohne Lücken abgeschlossen.
3. Ist es machbar, ein bakterielles Genom ausschließlich mit Plattformen für die Einzelmolekül-Sequenzierung der dritten Generation zu vervollständigen?
Nein, es ist nicht machbar. Kleine Plasmidfragmente (ungefähr 20 kb) können während des Bibliothekskonstruktionsprozesses verloren gehen. Darüber hinaus können bestimmte Regionen des Chromosoms aufgrund von Stichprobenwahrscheinlichkeitsproblemen oder Probenabbau nicht sequenziert werden.
4. Wie können wir die Genauigkeit der Assemblierung sicherstellen, angesichts der niedrigen Einzelbasisgenauigkeit von Plattformen der dritten Generation für die Einzelmolekülsequenzierung?
Die Einzelbasisgenauigkeit von Sequenzierungsdaten der dritten Generation liegt zwischen 87 % und 92 %. Um die Genauigkeit der Assemblierung sicherzustellen, können wir den folgenden dreistufigen Prozess anwenden:
- Korrigieren Sie die Sequenzierungsdaten vor der Assemblierung, indem Sie die Überlappung zwischen den Sequenzen der Einzelmolekül-Sequenzierung der dritten Generation nutzen.
- Nach der Assemblierung verwenden Sie Daten der dritten Generation der Einzelmolekülsequenzierung, um die assemblierten Sequenzen zu korrigieren.
- Nach der zweiten Korrektur verwenden Sie hochwertige zweite Generation. Hochdurchsatz-Sequenzierung Daten zur weiteren Korrektur der zusammengetragenen Ergebnisse.
Durch die Anwendung dieses dreistufigen Korrekturprozesses kann die endgültige Montagegenauigkeit 99,99 % übersteigen.
5. Wie geht die Langzeit-Sequenzierung mit repetitiven Regionen in bakteriellen Genomen um?
Die 15-25 kb langen erweiterten Leseweiten bieten eine einzigartige Lösung:
- Sie spannen effektiv und decken vollständig sich wiederholende Einheiten ab, wie IS-Elemente und rRNA-Cluster.
- Vermeiden Sie Zusammenbruch der Assemblierung, der häufig durch Kurzlese-Sequenzierung verursacht wird.
- Über 99% Vollständigkeit der Assemblierung in Bereichen mit sich wiederholenden Sequenzen nachgewiesen.
6. Ist ein separates Experiment für die epigenetische Detektion (6mA/4mC) erforderlich?
Nein, es sind keine zusätzlichen Experimente erforderlich. Mit der PacBio HiFi-Technologie:
- Basisänderungen werden nativ erfasst, ohne zusätzliche Bibliotheksvorbereitungen oder Sequenzierungsaufwände.
- Es bietet direkt eine umfassende Ganzgenom-Methylierungskarte.
- Empfindlichkeit: Erkennt Standorte mit einer Änderungsfrequenz von ≥85% mit über 95% Genauigkeit.
7. Beeinflusst abnormaler GC-Gehalt (<20% oder >80%) die Ergebnisse?
Die PacBio-Technologie neutralisiert GC-Bias:
- Stellt sicher, dass die Abdeckungsunterschiede in den 15-85% GC-Regionen unter 5% liegen.
- Keine Notwendigkeit für spezialisierte Bibliotheksoptimierung.
Bakterielle Gesamte Genom von neuem Seq Fallstudien
Kundenveröffentlichungshighlight
Phänotypische und Entwurf-Genomsequenzanalysen eines Paenibacillus sp. Isoliert aus dem Magen-Darm-Trakt eines nordamerikanischen Grauwolfs (Wolf)
Journal: Angewandte Mikrobiologie
Impact-Faktor: ~4,5 (2023)
Veröffentlicht: 23. September 2023
Hintergrund
Die canine entzündliche Darmerkrankung (cIBD) hat keine effektiven Behandlungen, wobei eine Dysbiose des Darms ein entscheidender Faktor ist. Graue Wölfe (Wolf), Vorfahren von Haushunden beherbergen einzigartige Darmmikrobiota, die möglicherweise während der Domestikation verloren gingen. Diese Studie isolierte ein sporenbildendes Paenibacillus sp. Stamm aus dem GI-Trakt eines wilden Wolfes, Charakterisierung seines probiotischen Potenzials zur Behandlung von cIBD.
Projektziele
- Isolieren & Phänotypisieren: Isolieren Sie chloroformresistente Sporenbildner aus dem Magen-Darm-Trakt von Wölfen; bewerten Sie die antimikrobielle Aktivität.
- Genomische Analyse: Sequenzieren und annotieren Sie das Genom, um probiotikabezogene Gene zu identifizieren.
- Phylogenetische Typisierung: Bestimmen Sie die taxonomische Identität und evolutionären Beziehungen.
CD Genomics Dienstleistungen
Als Partner für Genomik lieferte CD Genomics:
- Whole-Genome-Sequenzierung (WGS)
- Plattform: Illumina NovaSeq (400 Mbp Reads).
- Abdeckung: Entwurf der Assemblierung (7.034.206 bp).
- Bibliotheksvorbereitung: DNA-Extraktion.
- Bioinformatik Analyse
- Zusammenstellung & Annotation: JGI IMG/MER-Pipeline zur Genvorhersage (6.543 Gene).
- Funktionale Annotation: COG-Kategorisierung, Analyse konservierter Domänen (CD-Suche).
- Prophage-Erkennung: PHAge-Suchwerkzeug (PHASTER) für lysogene Sequenzen.
- Phylogenetische Analyse: BLAST+/MEGA für 16S rRNA-Typisierung; Mugsy/RAxML für die Phylogenie des gesamten Genoms.
Wichtigste Erkenntnisse
- Probiotischer Phänotyp validiert
- Antimikrobielle Aktivität: Gehemmt Staphylococcus aureus, Escherichia coli, und Micrococcus luteus (Abbildung 1B, Ergänzende Abbildung S1).
- Enzymproduktion: Stärkehydrolyse (Abbildung 1A), Lipase- und Cellulaseaktivität bestätigt.
- Sicherheitsprofil: Antibiotikaempfindlich (Tetracyclin, Erythromycin); keine Toxingene nachgewiesen.
- Genomische Einblicke durch die WGS von CD Genomics
- Antimikrobielle Gene: Bakteriocine (5), Lantibiotika (6), Chitinase (2), Lysin (22), Amidase (42) (Tabelle 1).
- Stoffwechselenzyme: Alpha-Amylase, Cellulase, Lipasen, Pektinase—entscheidend für die Kohlenhydratverdauung.
- Sporulation: 133 Gene für die Sporenbildung/Keimung (Verbesserung der Überlebensfähigkeit von Probiotika).
- Virale Elemente: 48 phagenabgeleitete Gene (nicht funktional, antimikrobielle Potenzial).
- Taxonomische Klassifikation
- 16S rRNA-Typisierung: 99% Identität zu Paenibacillus xylanexedens PAMC 22703.
- Phylogenomik: Nächste Verwandte: P. amylolyticus SQR-21 (dürreresistenter Weizenpartner) und Paenibacillus sp. OVF10 (medizinischer Pflanzenisolat) (Abbildung 3).
Zitierte Abbildungen
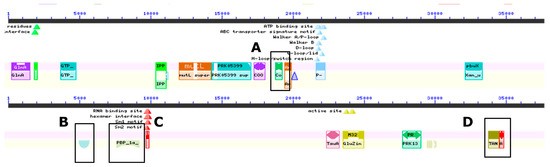 Abbildung 2. Konservierte Domänenanalyse: (A) Äußere Sporenhülle, (B) Sporulationsprotein K, (C) Penicillin-bindend, (D) Antibiotika-Synthese.
Abbildung 2. Konservierte Domänenanalyse: (A) Äußere Sporenhülle, (B) Sporulationsprotein K, (C) Penicillin-bindend, (D) Antibiotika-Synthese.
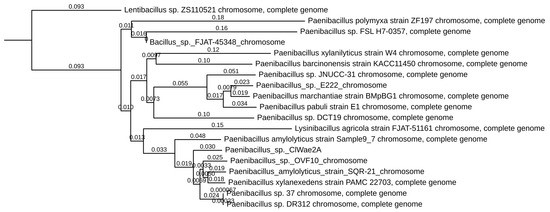 Abbildung 3. Phylogenetischer Baum von ClWae2A und verwandten Arten Paenibacillus spp. (Bootstrap >83%).
Abbildung 3. Phylogenetischer Baum von ClWae2A und verwandten Arten Paenibacillus spp. (Bootstrap >83%).
Implikationen
- Probiotische Entwicklung: ClWae2A’s Sporenbildung, Pathogenhemmung und kohlenhydratverdauende Enzyme positionieren es als Kandidaten für die Behandlung von IBD bei Hunden.
- Mikrobiom-Wiederherstellung: Die Wiederintroduktion von wolf-abgeleiteten Bakterien könnte Dysbiose, die durch Domestikation verursacht wurde, entgegenwirken.
- Präzisionsgenomik: Die WGS von CD Genomics ermöglichte die Identifizierung von Sicherheitsmarkern (keine Toxine) und funktionalen Genen (Antimikrobiellen/Enzymen), wodurch das Design von Probiotika weniger riskant wurde.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Identifizierung verschiedener Integron- und Plasmidstrukturen, die eine neuartige Carbapenemase bei Pseudomonas-Arten tragen
Zeitschrift: Front. Mikrobiol.
Jahr: 2019
Produktion eines bakteriozinenähnlichen Proteins PEG 446 aus Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotika und antimikrobielle Proteine
Jahr: 2024
Entwirrung der Rolle von Pathobionten aus Bacteroides-Arten bei entzündlichen Darmerkrankungen
Journal: bioRxiv
Jahr: 2023
Eine Chromosomenebene-Genomressource zur Untersuchung von Virulenzmechanismen und der Evolution des Kaffeerostpathogens Hemileia vastatrix
Journal: bioRxiv
Jahr: 2022
Streptomyces buecherae sp. nov., ein Actinomycet, der aus mehreren Fledermausarten isoliert wurde.
Journal: Antonie van Leeuwenhoek
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


