Oxford Nanopore Sequenzierungsdienste — Ultra-lange Reads, Echtzeitentscheidungen, direkte RNA
Die einzige Sequenzierungstechnologie, die Mb-Klasse ultra-lange Reads liefert, Daten in Echtzeit streamt und native RNA direkt sequenziert — ohne Reverse Transkription oder Amplifikation. Die Oxford Nanopore-Plattform von CD Genomics bietet Ihnen die längsten Reads in der Genomik, die Möglichkeit, einen Lauf zu stoppen, wenn Sie genügend Daten haben, und direkten Zugang zu RNA-Modifikationen, die andere Plattformen übersehen.
Was wir anbieten:
Ultra-lange Reads (Mb-Klasse) für lückenfreie Genomassemblierung und komplexe Wiederholungsauflösung
Direkte RNA-Sequenzierung — native RNA, keine reversen Transkription, keine Amplifikationsverzerrung
Echtzeit-Sequenzierungssteuerung — überwachen Sie den Ertrag live, stoppen Sie, wenn die Ziele erreicht sind.
Wenn Ihr Projekt Lesevorgänge erfordert, die länger als 20 kb sind – um große strukturelle Varianten abzudecken, Genomlücken zu schließen oder vollständige Transkript-Isoformen zu entschlüsseln – ist Oxford Nanopore Ihre beste Wahl. Nanopore hält den Rekord für die längsten jemals produzierten Sequenzierungslese (über 2 Mb). Es ist auch die einzige Plattform, die Daten in Echtzeit streamt – sodass Sie einen Lauf sofort stoppen können, sobald Sie genügend Abdeckung haben – und die einzige Plattform, die native RNA-Moleküle direkt sequenziert und dabei Modifikationsinformationen bewahrt, die cDNA-basierte Methoden verlieren.
Natürlich tauschen rohe Nanoporen-Lesungen eine gewisse Genauigkeit pro Lesung gegen diese Fähigkeiten ein (typischerweise Q10–20 roh, verbessernd mit der neuesten Chemie und Dorado-Basecallern). Für Projekte, die die höchste Genauigkeit pro Lesung erfordern, sollten Sie PacBio HiFi in Betracht ziehen; für die längsten Lesespannen, die Echtzeitüberwachung und die direkte RNA-Analyse hat Nanopore keine Konkurrenz.
Wie Nanoporen-Sequenzierung funktioniert
Nanopore-Sequenzierung lässt ein einzelnes DNA- oder RNA-Molekül durch einen in eine elektrisch resistente Membran eingebetteten Protein-Nanopore passieren. Während jedes Nukleotid den Pore durchquert, stört es den ionischen Strom auf eine sequenzspezifische Weise. Diese Stromänderungen werden in Echtzeit aufgezeichnet und von einem neuronalen Netzwerk-Basiscaller wie Dorado in Nukleotidsequenzen (A, T, C, G — oder RNA-Basen) decodiert.
Im Gegensatz zu Illumina (Sequenzierung durch Synthese mit Clusteramplifikation) oder PacBio (optische Erkennung von fluoreszenzmarkierten Nukleotiden in Zero-Mode-Wellenleitern mit zirkulärem Konsens) liest Nanopore das native Molekül direkt. Es sind keine Amplifikation, keine Synthese und keine optischen Messungen erforderlich. Das Rohsignal trägt nicht nur die Basenidentität, sondern auch Modifikationsinformationen (5mC, 6mA und RNA-Modifikationen), die bioinformatisch ohne zusätzliche Probenvorbereitung extrahiert werden können.
Warum es für Ihre Forschung wichtig ist:
Ultra-lange Reads (die routinemäßig 1 Mb überschreiten, mit Rekord-Reads, die 2 Mb übersteigen). Sie umfassen Telomere, Zentromere, segmentale Duplikationen und große strukturelle Varianten, die mit Plattformen für kurze oder mittellange Reads nicht aufgelöst werden können.
Echtzeit-Datenstreaming. Sequenzierungsergebnisse werden erzeugt, während das Molekül durch die Pore passiert, was eine Live-Überwachung der Ausbeute, adaptive Probenahme (Zielregionen in Echtzeit anreichern oder reduzieren) und eine sofortige Beendigung des Laufs ermöglicht, wenn genügend Daten gesammelt wurden.
Direkte RNA- und native Modifikationsdetektion. Sequenzieren Sie RNA-Moleküle ohne Reverse-Transkription oder Amplifikationsverzerrung. Erkennen Sie DNA-Methylierung (5mC, 6mA) aus genomischen Reads und RNA-Modifikationen (m6A, Pseudouridin, Inosin) aus direkten RNA-Reads – alles aus dem Rohsignal, ohne dass eine Bisulfit-Konversion oder Anreicherungsschritte erforderlich sind.
EinzeilerVollständige tRNA-Sequenzierung mit direkter RNA-Modifikationsdetektion auf der Nanopore-Plattform. Umfassende Profilierung von tRNA-Isoacceptoren und Isoformen mit Erfassung von nativen Modifikationen, einschließlich m1A, m3C, m7G, Pseudouridin und anderen epitranskriptomischen Markierungen.
Am besten fürtRNA-Biologie, Studien zur Modifikationslandschaft, tsRNA-Charakterisierung, epitranskriptomisches Profiling.
EinzeilerLösen Sie komplexe mikrobielle Gemeinschaften mit langen Nanoporen-Lesungen für die Klassifizierung auf Artenebene, metagenomisch assemblierte Genome (MAGs) und Profilierung von Resistenzgenen. Verfügbar auf den Plattformen PacBio HiFi und Oxford Nanopore.
Am besten fürKomplexe metagenomische Gemeinschaften, vor Ort einsetzbare Metagenomik, Echtzeit-Pathogenüberwachung.
EinzeilerSequenzieren Sie vollständige cDNA-Moleküle für die Entdeckung von Transkripten auf Isoform-Ebene. Transkribieren Sie RNA in cDNA und sequenzieren Sie dann direkt – erfassen Sie vollständige Transkriptstrukturen vom 5'-Ende bis zum 3'-Ende ohne Assemblierung.
Am besten fürVollständige Transkript-Isoform-Entdeckung, Identifizierung neuer Transkripte, Quantifizierung der Isoform-Expression.
EinzeiligAmplifizieren und sequenzieren Sie vollständige rRNA-Genregionen (16S, 18S, ITS) mit Nanopore-Langlesen für eine Arten- und Stammebene taxonomische Auflösung, die mit Kurzlese-Ampliconen nicht erreicht werden kann.
Am besten fürMikrobielle Gemeinschaftsprofilierung, Analyse des Umweltmikrobioms, taxonomische Klassifikation auf Stammebene.
Was Sie erhalten werden
Jedes Oxford Nanopore-Projekt wird mit transparenten Daten, detaillierter Dokumentation und reproduzierbaren QC-Metriken geliefert – für ein publikationsreifes Vertrauen.
Kern-Daten Dateien (für jedes Projekt)
FASTQ (.fastq.gz) — basierte Reads (Dorado).
Optionales Rohsignal — FAST5/POD5 auf Anfrage für modifikationsbewusste Neuanalyse.
Projektmemo – das verwendete Nanoporen-Sequenzierungsprotokoll (Bibliothekskit, Flusszelle/Chemie, Laufparameter), sowie Softwareversionen und -parameter.
Eingang & QualitätskontrolleQubit quant; A260/280 ~1,8–2,0, A260/230 ≥2,0. HMW gDNA für Ultra-Long; RNA RIN ≥8.
Bibliotheksvorbereitung (nach Anwendung)Standard WGS; Ultra-lang (sanfte HMW-Handhabung); Amplicon; Zielgerichtet (Cas9 oder adaptive Probenahme); Vollständiges cDNA/lncRNA; Direkte RNA; Pore-C; TAIL Iso-Seq.
Flusszelle & Barcodes: Größen, um Erträge/Ziele zu erzielen; Ausgleich von barcode-gestützten Eingaben.
Sequenzierung & Steuerung1–72 h typisch; Live-Überwachung; stoppen/verlängern oder neu laden nach Bedarf; adaptive Probenahme aktivieren, wo unterstützt.
Bereichern Sie schnell Loci von Interesse, ohne die Kosten einer Vollgenomsequenzierung.
Empfohlene Dienstleistungen: Gezielte Nanoporen-Sequenzierung (Cas9 oder adaptive Probenahme), Amplicon-Sequenzierung.
3D-Genomarchitektur
Erstellen Sie Langstrecken-Kontaktkarten für Scaffolding- und Chromatinstudien.
Empfohlener Service: Pore-C.
Metagenomik & Pathogenüberwachung
Verbessern Sie die Montage-/Belastungsauflösung; profitieren Sie von Echtzeitentscheidungen.
Empfohlene Dienstleistungen: Standard WGS, gezielt, Amplicon (pro Design).
Für hochauflösende Kohorten mit kleinen Varianten können Kurzleseverfahren kosteneffizient sein; hybride Designs (Kurzlese + Nanopore) erfassen sowohl die Tiefe als auch den langfristigen Kontext.
Nanopore vs PacBio HiFi vs Illumina — Plattformvergleich
Die richtige Plattform für Ihr Projekt auswählen? Hier ist, wie Nanopore im Vergleich zu PacBio HiFi und Illumina in den Dimensionen abschneidet, die für Forschungsergebnisse wichtig sind.
Dimension
Nanopore (ONT)
PacBio HiFi
Illumina
Prinzip
Ionenstrom durch ein Protein-Nanopore; neuronales Netzwerk zur Basisbestimmung
Optische Detektion in ZMWs; zirkulärer Konsens (CCS)
Sequenzierung durch Synthese; Cluster-Bildgebung
Leseumfang (typisch)
10–100 kb Routine; ultra-lang bis 2 Mb+
~15–20 kb HiFi-Lesungen; Subreads bis zu ~100 kb
Bis zu 2×300 bp
Vorlesegenauigkeit (roh)
Q10–Q20 Rohdaten; Verbesserung mit R10.4.1 + Dorado; hohe Genauigkeit mit Konsens-Tiefe
QV ≥30 (≥99,9%) gemäß CCS-Konsens
≥99,9% roh
Datenzeitpunkt
Echtzeit-Streaming; einen Lauf live stoppen oder verlängern
Batch (Analyse nach Abschluss des Laufs)
Charge
Einheimische Biologie
Direkte RNA-Sequenzierung; 5mC/6mA aus Rohsignal; RNA-Modifikationen ohne RT oder Amplifikation
5mC aus der Polymerase-Kinetik; kein Bisulfit erforderlich
Keine native Änderungsdetektion (Standard-Workflows)
Wo es glänzt
Ultra-lange Spannweite (Telomere, massive SVs, Lückenverschluss); Echtzeit-/Feldarbeit; Direkte RNA
Höchste Genauigkeit bei Langlesungen; Assemblierungen und Methylierung aus einem Datensatz
Tiefe SNV/Indel-Kohorten; kosteneffiziente große Studien
Abwägungen
Rohgenauigkeit niedriger als HiFi oder Illumina; signalbewusste Bioinformatik erforderlich
Längere Laufzeiten; keine Echtzeitsteuerung oder Streaming
Kein langfristiger Kontext; kann native Änderungen nicht erkennen.
Schnelle Entscheidungsanleitung:
Benötigt Reads länger als 100 kb oder feldverfügbare Sequenzierung → Nanopore.
Benötigen Sie die höchste Genauigkeit bei langen Reads für Assemblierungen und Variantenaufrufe → PacBio HiFi.
Für direkte RNA-Sequenzierung oder Echtzeitdaten ist Nanopore die einzige Option.
Große SNV/Indel-Kohorten mit einem Budget → Illumina für Tiefe; Nanopore-Langlesungen für SVs und Phasierung hinzufügen.
Hybride Designs: Nanopore-Ultra-Long + PacBio HiFi für vollständige T2T-Assemblierungen; Nanopore-Langreads + Illumina-Kurzreads für SV-Erkennung mit tiefgehender SNV-Validierung.
Die tatsächliche Leistung variiert je nach Probenqualität, Bibliotheksvorbereitung, Sequenzierungstiefe und Analysepipeline.
Qualität & Studiendesign
Replikate & KontrollenEmpfehlen Sie ≥2 biologische Replikate pro Bedingung; negative Kontrollen für Amplicon/Targeted einbeziehen; optionale Spike-Ins für Methylierung.
AbdeckungsplanungGröße der Long-Read-Tiefe in Bezug auf die Genomkomplexität/SV-Ziele; planen Sie die On-Target-Tiefe für gezielte/Amplikon-Analysen; Größe der Reads/Probe für Isoformen oder direktes RNA.
AkzeptanzkriterienVereinbarte Ziele für Ertrag pro Barcode, Zielanteil, Mapping-Rate und Leseprofil (z. B. N50-Ziele für Ultra-Long).
LaufzeitentscheidungenNutzen Sie Echtzeit-Dashboards, um effizient zu stoppen, zu verlängern oder neu zu laden; dokumentieren Sie alle Abweichungen im Projektprotokoll.
Der CD Genomics Vorteil
1
Anwendungsorientierte Abgrenzung
Ordnen Sie Ihre biologische Frage dem richtigen Oxford Nanopore-Service zu (Ultra-Long, Direct RNA, Full-Length Transcript/lncRNA, Targeted/Cas9 oder adaptiv, Amplicon, Pore-C, TAIL Iso-Seq).
2
Entwurfsmodellierung & Machbarkeit
Abdeckungsmodellierung, Barcode-Balance und Machbarkeitsprüfungen von Ziel/Primer, bevor Sie sich festlegen.
3
Reproduzierbare Analytik
Containerisierte/versionsgesperrte Pipelines; saubere Ergebnisverpackung mit Analysebericht und Datenwörterbuch.
4
Echtzeit-Lauf-Effizienz
Live-Dashboards zum Stoppen/Erweitern/Neuladen, wenn Ziele erreicht werden; adaptive Stichprobenahme, wenn geeignet.
5
Datenintegrität und -sicherheit
Strukturierte Ordner, Prüfziffernüberprüfung und gesicherter Transfer mit einer festgelegten Aufbewahrungsrichtlinie.
6
Veröffentlichungsreife Unterstützung
Optionale Ergebnisse-Durchführung, Vorbereitung von Abbildungen/Tabellen und Unterstützung bei Manuskripten/Überprüfungen.
7
Plattformübergreifende Neutralität
Zielgerichtete Anleitung, wann hybride Strategien (Short-Read + ONT oder HiFi + ONT) einen Mehrwert bieten.
Musteranforderungen
Dienstleistung
Menge & Integrität
Reinheit
Lagerung / Versand
Wichtige Hinweise
WGS (Standard)
≥1–3 μg gDNA, ≥30 kb
A260/280 1,8–2,0; A260/230 ≥2,0
−20 °C auf Eis
Bereitstellung der Extraktionsmethode; kein Vortexen
Ultra-lange WGS
≥3–5 μg HMW gDNA, ≥50 kb bevorzugt
A260/280 1,8–2,0; A260/230 ≥2,0
−20 °C
Sanfte Handhabung ist entscheidend; nur Weitwinkelspitzen
Direkte RNA-Sequenzierung
≥500 ng–1 μg poly(A)+ RNA; RIN ≥7
A260/280 ~2,0; A260/230 ≥2,0
−80 °C Trockeneis
Nicht erhitzen oder denaturieren vor dem Versand.
Vollständiges Transkript
≥500 ng–1 μg Gesamt-RNA; RIN ≥8
A260/280 ~2,0; A260/230 ≥2,0
−80 °C Trockeneis
Poly(A)+ oder rRNA-unterdrückt nach Design
Vollständige LncRNA
≥500 ng–1 μg Gesamt-RNA; RIN ≥7
Das Gleiche wie oben
−80 °C Trockeneis
rRNA-Depletion oder Poly(A)-Selektion
Amplicon-Sequenzierung
≥50–200 ng gepoolte Amplicons
Saubere PCR; keine Primer-Dimere
4°C Kältepackung
Bitte geben Sie die Primer-Tabelle und das Zielgen an.
Gezielt (Cas9/adaptiv)
≥1–3 μg gDNA
A260/280 1,8–2,0
−20 °C
Bitte stellen Sie eine BED-Datei für die Zielregion oder eine Genliste zur Verfügung.
Pore-C
≥1–2 μg HMW gDNA, ≥50 kb
A260/280 1,8–2,0
−20 °C
Sanfte Handhabung; kein Vortexing
TAIL Iso-Seq
≥500 ng–1 μg Gesamt-RNA; RIN ≥7
A260/280 ~2,0
−80 °C Trockeneis
Poly(A)+-Selektion erforderlich
Nano tRNA-Sequenzierung
≥500 ng Gesamt-RNA; RIN ≥7
A260/280 ~2,0; A260/230 ≥2,0
−80 °C Trockeneis
Bitte geben Sie die tRNA-Anreicherungsmethode an, wenn sie selbst angereichert ist.
Lange Lesungen Amplicon
≥100 ng gepoolte Amplicons; ≥500 bp Ziel
Saubere PCR; keine Primer-Dimere
4°C Kältepackung
Bereitstellen von Primer-Tabelle + Zielkoordinaten
Allgemeine Hinweise
Vermeiden Sie Inhibitoren (Phenol, Heparin, EDTA, Polysaccharide) und wiederholtes Einfrieren und Auftauen; trocknen Sie die Perlen nicht zu stark.
Verwenden Sie breite Spitzen und kein Vortexen für HMW-DNA.
Fügen Sie eine kurze Extraktionsmethode und bekannte Verunreinigungen hinzu.
Niedrig-input/FFPE oder ungewöhnliche Matrizen könnten machbar sein – kontaktieren Sie uns für ein maßgeschneidertes Protokoll.
Autoren: Bin Sun, Kaushal Kumar Bhati, Peizhe Song u.a.
Veröffentlichungsdatum: 27. September 2022
Journal: PLOS Genetik
Forschungsfrage (Aufmerksamkeit):
Welches Enzym installiert m^6A am 3′UTR der mRNA des FLOWERING LOCUS C (FLC), und wie beeinflusst diese Modifikation die Stabilität des FLC-Transkripts und die Blüte?
Ansatz:
Ein Multi-Omics-Design integrierte die Oxford Nanopore Direct RNA-Sequenzierung, mRNA-seq und meRIP-seq, um die differentielle Expression und die differentielle RNA-Methylierung in Wildtyp- vs. FIONA1 (FIO1)-Mutantenpflanzen zu profilieren. Die Direct RNA erfasste native Signalmerkmale, während meRIP-seq m^6A-reiche Regionen kartierte; die kombinierten Beweise identifizierten die Modifikationsstelle im 3′UTR von FLC.
Wichtigste Erkenntnisse:
FIO1 ist die Methyltransferase, die für die m^6A-Modifikation im 3′UTR von FLC-mRNA verantwortlich ist.
Der Verlust dieser 3'-End-Methylierung in fio1-Mutanten destabilisiert das FLC-mRNA (reduzierte FLC-Spiegel) und trägt zu einem frühblühenden Phänotyp bei.
Die Autoren veröffentlichten Nanopore-Direkt-RNA-Daten (GEO: GSE212766) und passende mRNA/meRIP-Datensätze, die die Reproduzierbarkeit unterstützen.
Direkte RNA-Sequenzierungsanalyse.
Was dies demonstriert:
Diese peer-reviewed Studie zeigt, wie die Nanopore-Direkt-RNA-Sequenzierung – kombiniert mit der Analyse von RNA-Modifikationen (m^6A) – biologische Fragen beantwortet, die von nativer RNA und post-transkriptionaler Regulation abhängen. Sie ist ein starkes Beispiel für "Nanopore-Sequenzierungstechnologie, Bioinformatik und Anwendungen" in der Epitranskriptomik und den genregulatorischen Mechanismen.
Wie CD Genomics ein ähnliches Projekt abstecken würde.:
DienstleistungNanopore Direkte RNA-Sequenzierung (+ optionale meRIP-seq über Partner-Workflow)
AnalyseIsoform-Profilierung, modifikationsassoziierte Signalsummen, differentielle Expression, Integration mit immunpräzipitationsbasierten m^6A-Spitzen
LiefergegenständeFASTQ (Direktes RNA), optional POD5/FAST5, QC-Bericht, Zusammenfassungen der Modifikationen, Abbildungen/Tabellen geeignet für die Veröffentlichung
Häufig gestellte Fragen — Nanopore-Sequenzierung
Nur für Forschungszwecke, nicht zur klinischen Diagnose, Behandlung oder individuellen Gesundheitsbewertung bestimmt.
Veröffentlichungen
FIONA1-mediated methylation of the 3’UTR of FLC affects FLC transcript levels and flowering in Arabidopsis

 Richtlinien zur Einreichung von Proben
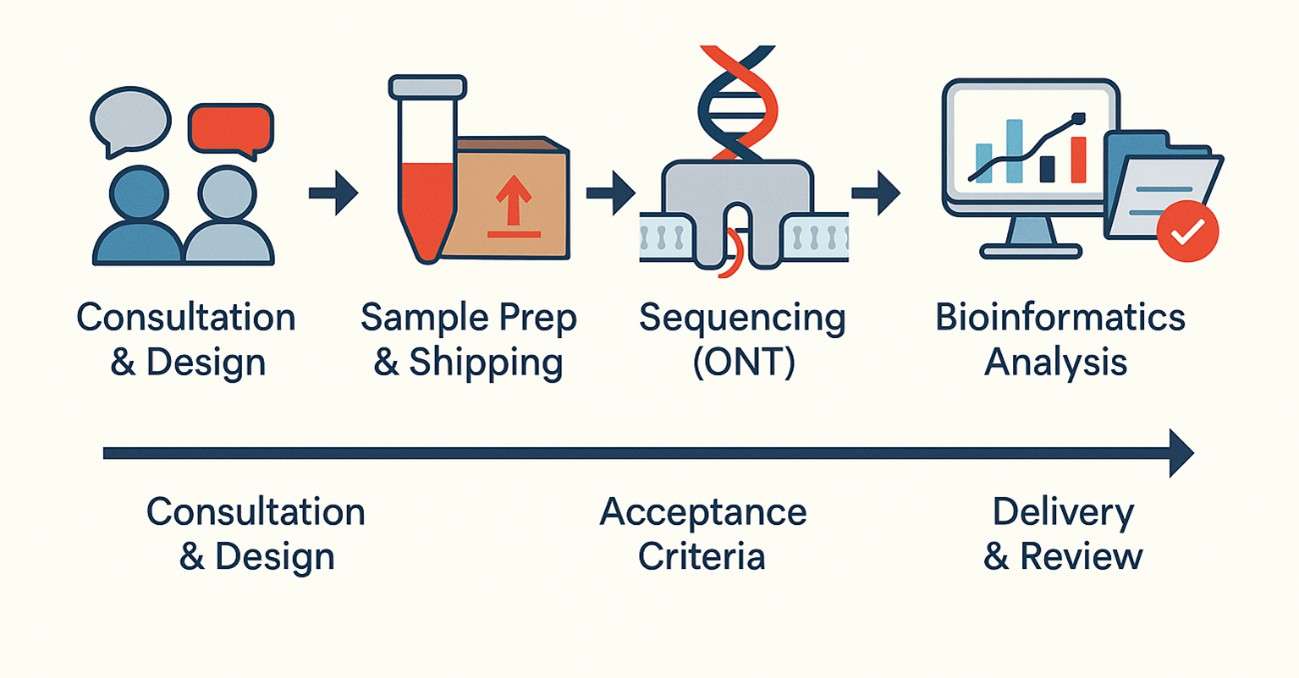
Richtlinien zur Einreichung von Proben



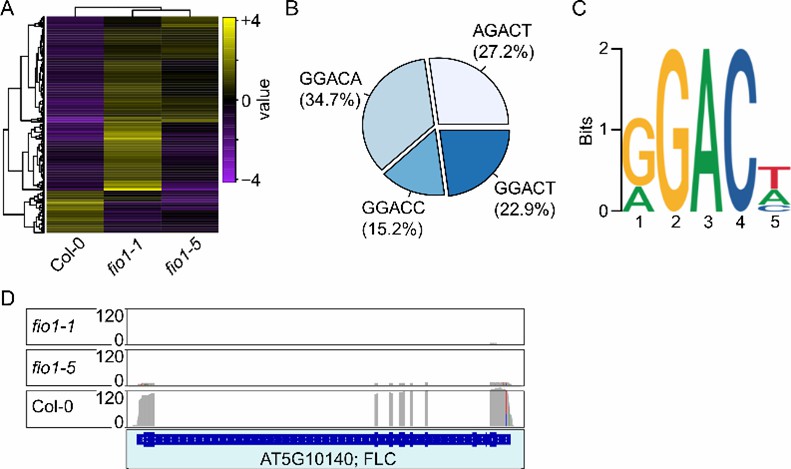 Direkte RNA-Sequenzierungsanalyse.
Direkte RNA-Sequenzierungsanalyse.

