
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben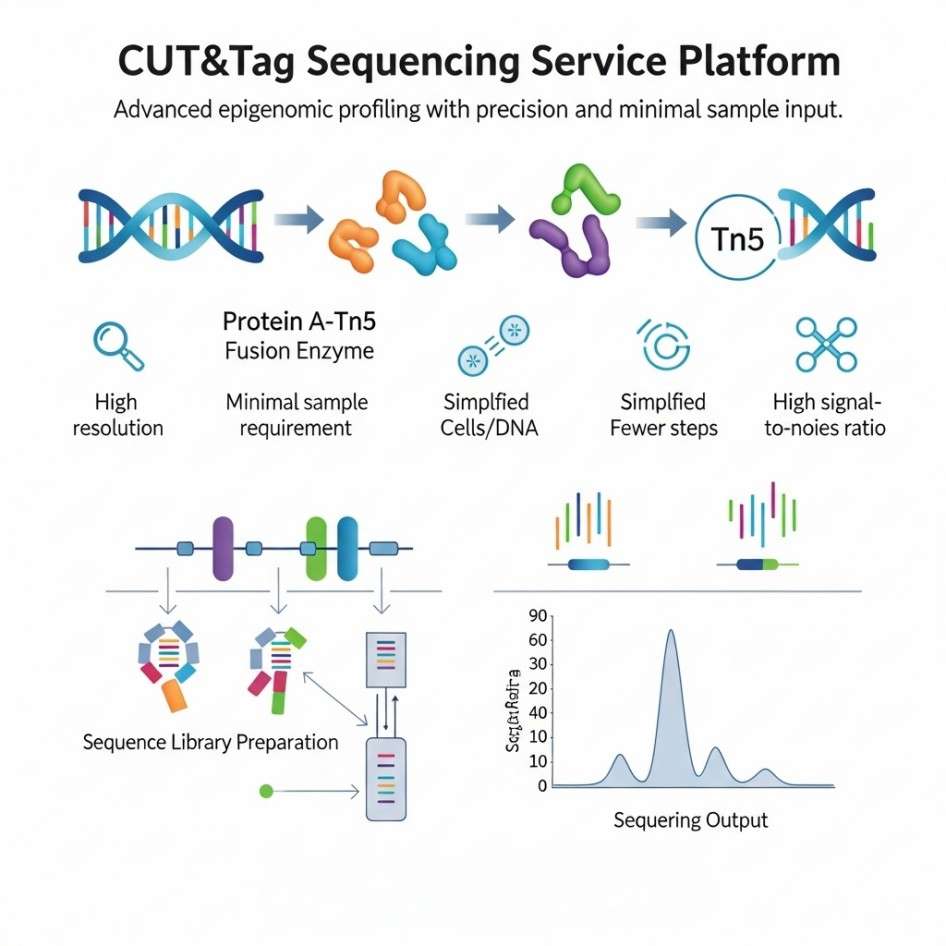
Was ist CUT&Tag-Sequenzierung?
CUT&Tag-Sequenzierung (Cleavage Under Target und Tagmentation) ist eine revolutionäre Methode zur Untersuchung von Protein-DNA-Interaktionen und Chromatinstruktur. Diese Technik ermöglicht eine präzise Profilierung von DNA-assoziierten Proteinen, einschließlich Transkriptionsfaktoren und Histonen, und benötigt dabei nur minimale Mengen an Probenmaterial. CUT&Tag-Sequenzierung wird als bahnbrechend angesehen, da sie die Fähigkeit besitzt, hochauflösende Daten mit reduziertem Hintergrundrauschen im Vergleich zu traditionellen Sequenzierungsmethoden bereitzustellen.
Der Prozess beginnt mit der Verwendung eines spezifischen Antikörpers, um ein Protein von Interesse zu zielen, wie beispielsweise einen Transkriptionsfaktor oder eine Histonmodifikation. Sobald der Antikörper an das Ziel bindet, wird ein Tagging-Enzym verwendet, um die DNA in unmittelbarer Nähe zum Protein zu markieren. Dies ermöglicht es den Forschern, die genauen Regionen des Genoms zu identifizieren, an denen das Protein mit der DNA interagiert. Nach diesem Tagging-Schritt wird eine Sequenzierung durchgeführt, um eine detaillierte Karte der Bindungsstellen zu erstellen.
Die Hauptvorteile von CUT&Tag-Analyse liegt in seiner Effizienz und Sensitivität. Traditionelle Techniken wie ChIP-seq erfordern oft große Mengen an Ausgangsmaterial und zeitaufwändigere Protokolle. Im Gegensatz dazu benötigt die CUT&Tag-Sequenzierung deutlich weniger Zellen, was die Komplexität und die Kosten der Probenvorbereitung reduziert. Darüber hinaus liefert die CUT&Tag-Sequenzierung hochspezifische Ergebnisse mit weniger Hintergrundrauschen, was den Forschern klarere Einblicke in Protein-DNA-Interaktionen bietet.
Die Fähigkeit dieser Methode, hochwertige Daten mit weniger Ressourcen bereitzustellen, macht sie zu einer attraktiven Option für verschiedene Genomik- und Epigenomik-Studien, insbesondere bei der Arbeit mit begrenzten Stichprobengrößen oder seltenen Zellpopulationen.
✅ Wichtige Überlegungen:
- CUT&Tag benötigt deutlich weniger Zellen als ChIP-seq, was es ideal für Studien mit begrenztem Probenmaterial macht.
- Zellfixierung: Im Gegensatz zu ChIP-seq erfordert CUT&Tag keine Zellfixierung, was den Arbeitsablauf vereinfacht und potenzielle Artefakte reduziert.
- Chromatinfragmentierung: CUT&Tag nutzt Tn5-Transposase für eine präzise DNA-Fragmentierung, während ChIP-seq häufig MNase oder Sonikation verwendet, was Variabilität einführen kann.
- Sequenzierungstiefe: ATAC-seq benötigt typischerweise weniger Sequenzierungsreads im Vergleich zu ChIP-seq, was es für bestimmte Anwendungen kostengünstiger macht.
- Bibliothekskonstruktion: Die direkte Ligationstechnik von CUT&Tag vereinfacht die Bibliotheksvorbereitung, während ChIP-seq zusätzliche Schritte umfasst, die die Komplexität und den Zeitaufwand erhöhen können.
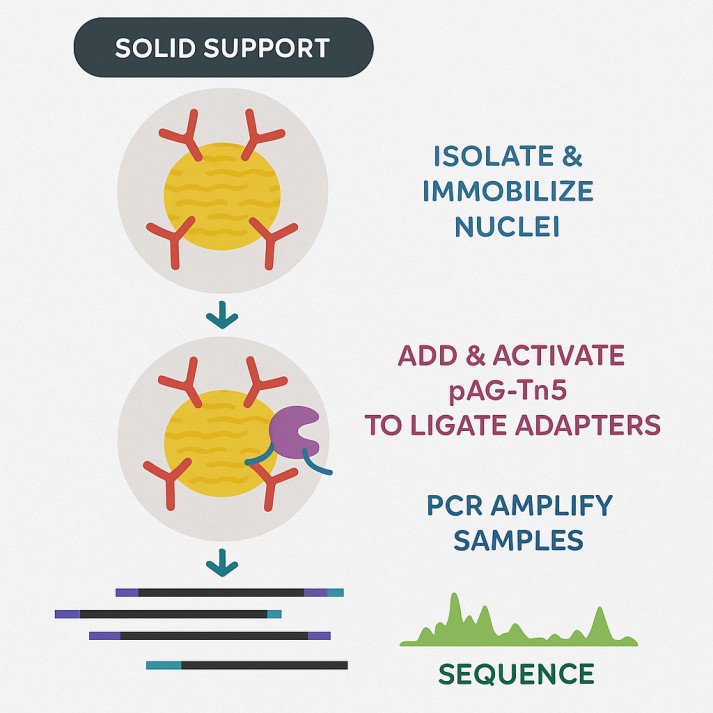
Experimenteller Workflow für CUT&Tag-Sequenzierung
- ZellvorbereitungBeginnen Sie mit der Vorbereitung einer Zellaufhängung aus Gewebe- oder Zellproben und bewerten Sie die Zellviabilität.
- Primäre AntikörperbindungFügen Sie den primären Antikörper hinzu, um das Zielprotein zu markieren.
- Sekundäre AntikörperbindungDer sekundäre Antikörper bindet an den primären Antikörper und bildet einen Komplex mit dem Zielprotein.
- Protein A-Tn5-Fusionsproteinbindung und DNA-FragmenteDas Protein A-Tn5-Fusionsprotein bindet an das antikörpergerichtete Protein, schneidet die nahegelegene DNA und fügt Sequenzierungsadapter hinzu.
- DNA-Fragmentreinigung und BibliothekskonstruktionReinigen Sie die fragmentierte DNA und erstellen Sie die Sequenzierungsbibliothek.
- Sequenzierung und DatenanalyseFühren Sie Hochdurchsatz-Sequenzierung durch und führen Sie eine umfassende Datenanalyse durch.
- ErgebnisberichtEin detaillierter Bericht über die Ergebnisse wird zur weiteren Interpretation bereitgestellt.
Vorteile von CUT&Tag gegenüber anderen Sequenzierungsmethoden
Niedriger ZelleneingangBereits 60 Zellen können für genaue Ergebnisse verwendet werden.
Vereinfachter ProzessKeine Notwendigkeit für Cross-Linking, Chromatin-Sonikation oder Immunpräzipitation (IP). Dies reduziert die Komplexität des Workflows.
Hohe Signal-Rausch-VerhältnisDie Methode erzeugt sauberere Daten mit geringem Hintergrundrauschen, wodurch die Notwendigkeit chemischer Quervernetzungen entfällt und nicht-spezifische Bindungen reduziert werden.
Ausgezeichnete ReproduzierbarkeitDie Ergebnisse sind äußerst konsistent, und es besteht keine Notwendigkeit für Eingabekorrekturen.
Kürzerer ExperimentierzyklusDie Integration von Fragmentierung und Bibliothekskonstruktion verkürzt den gesamten Experimentzeitraum erheblich.
Hohe Auflösung, Empfindlichkeit und ZuverlässigkeitDiese Technik ermöglicht eine präzise Kartierung von Bindungsstellen für Transkriptionsfaktoren und Histonmodifikationen.
Umfassende bioinformatische Unterstützung
Bewertung der Datenqualität von Sequenzierungen
- Genom-Ausrichtungsanalyse: Bewerten Sie die Qualität und Genauigkeit der Genom-Ausrichtung und geben Sie Einblicke in die Zuverlässigkeit der Sequenzierung.
- Genomverteilungsstatistiken: Analysieren Sie die Verteilung der zugeordneten Reads im gesamten Genom, um ein besseres Verständnis der Sequenzierungstiefe und -abdeckung zu erhalten.
- Peak-Calling-Analyse und Grundlegende Statistiken: Identifizieren Sie Regionen von Interesse (Peaks) im Genom und berechnen Sie grundlegende Statistiken wie Peak-Häufigkeit und -Standort.
- Spitzenverteilung in genomischen Elementen: Karte, wo identifizierte Spitzen in verschiedenen genomischen Elementen auftreten, wie z.B. Promotoren oder Enhancern.
- Motivanalyse von Transkriptionsbindungsstellen: Analysieren Sie nur für Transkriptionsfaktoren die Bindungsmotive innerhalb der identifizierten Peaks und bieten Sie Einblicke in die Genregulation.
Genfunktionannotation für Spitzenziele
- Gene-Ontologie (GO) Analyse: Identifizieren Sie biologische Prozesse, molekulare Funktionen und zelluläre Komponenten, die mit den Zielgenen der Spitzenwerte verbunden sind.
- Weganalyse: Ordnen Sie die Spitzenzielgene relevanten biologischen Wegen zu, um ihre Rolle in zellulären Prozessen zu entdecken.
- Differenzielle Spitzenanalyse: Vergleichen Sie Spitzen unter verschiedenen Bedingungen, um signifikante Veränderungen in genomischen Merkmalen zu identifizieren.
- Integration mit anderen Omics-Daten: Kombinieren Sie die Ergebnisse der Peak-Analyse mit anderen Omics-Daten (z. B. Transkriptomik), um ein tieferes Verständnis der Genregulationsnetzwerke zu erlangen.
Anwendungen der CUT&Tag-Sequenzierung in der Genomik und Epigenomik
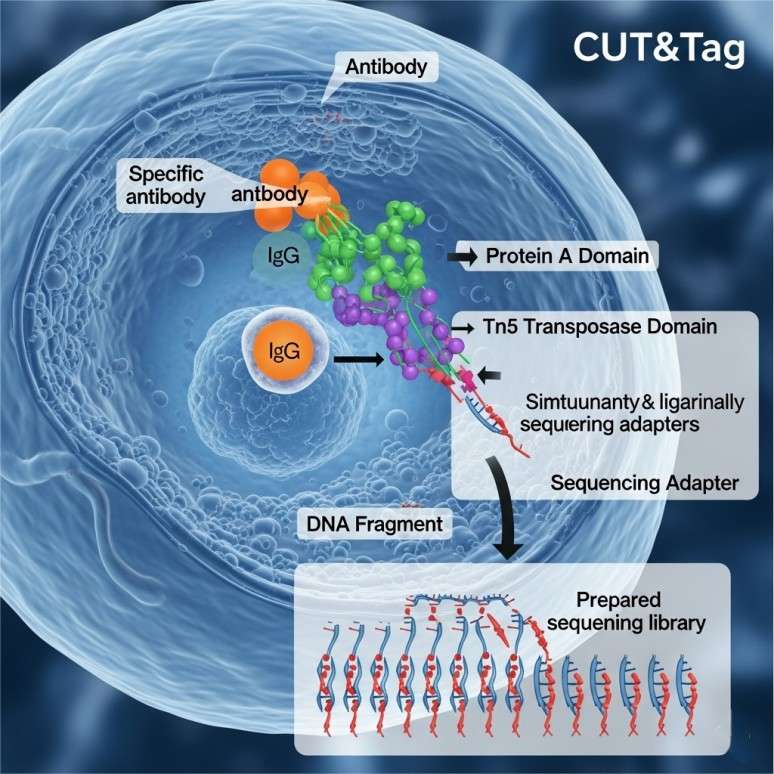
Enhancer-Kartierung und Super-Enhancer-Identifizierung (H3K27ac)
- Zweck: CUT&Tag wird verwendet, um Enhancer-Regionen, einschließlich Super-Enhancer, zu kartieren, die für die Genaktivierung entscheidend sind.
- Anwendung: Dies hilft, wichtige regulatorische Elemente zu identifizieren, die die Genexpression modulieren, und bietet ein klareres Verständnis der genetischen Regulationsnetzwerke.
Aktive Promotorregionen (H3K4me3)
- Zweck: Konzentriert sich auf Transkriptionsstartstellen und identifiziert aktive Promotorregionen, die an der Initiierung der Genexpression beteiligt sind.
- Anwendung: Vital für das Verständnis, wie Genpromotoren die Transkription auslösen und die Mechanismen der Genaktivierung aufdecken.
Histonmodifikationen (Lactylierung und Butyrylierung)
- Zweck: CUT&Tag kann Histonlactylierung und -butyrylierung nachweisen, Modifikationen, die eng mit aktiver Genexpression verbunden sind.
- Anwendung:
- Histon-Lactylierung: An Genpromotoren angereichert, verbunden mit Prozessen wie Angiogenese und Makrophagenpolarisation.
- Histonbutyrylierung: Entscheidend für die Regulierung der Genexpression und beteiligt an biologischen Prozessen wie RNA-Spleißen, Proteinsynthese und DNA-Reparatur.
DNA G-Quadruplexe (G4)
- Zweck: Identifiziert G4-Strukturen in Gen-Promotoren, 5' UTRs und Telomeren, die die DNA-Replikation und Transkription beeinflussen.
- Anwendung: G4-Strukturen fungieren als Regulatoren der Genexpression und sind mit Krankheiten wie Krebs und neurodegenerativen Erkrankungen assoziiert.
Identifizierung von Transkriptionsfaktor-Bindungsstellen
- Zweck: CUT&Tag identifiziert präzise die Bindungsstellen von Transkriptionsfaktoren und liefert Einblicke in die Genregulation.
- Anwendung: Verbessert unser Verständnis darüber, wie Transkriptionsfaktoren die Genexpression steuern, indem sie ihre spezifischen Bindungsstellen auf der DNA identifizieren.
Analyse der Histonmodifikation
- Zweck: Analysiert Histonmodifikationen mit hoher Auflösung, um zu verstehen, wie diese Modifikationen die Genaktivierung oder -stilllegung regulieren.
- Anwendung: Hilft dabei, zu erkunden, wie Histonmodifikationen zwischen Zelltypen oder Entwicklungsstadien variieren, und bietet Einblicke in die Regulation der Genexpression und die Mechanismen von Krankheiten.
Chromatin-Zugänglichkeit und Genregulatorische Netzwerke
- Zweck: Erkennt Regionen offenen Chromatins, die eng mit der Gentranskription verbunden sind.
- Anwendung: Unterstützt den Aufbau von Genregulationsnetzwerken, die für das Verständnis der Genexpression entscheidend sind und neue Einblicke in Krankheitsmechanismen bieten.
Organoid epigenetische Regulation
- Zweck: Untersucht die epigenetische Regulation während der Organoidentwicklung, die entscheidend für das Verständnis von Krankheitsmechanismen ist.
- Anwendung: Bietet neue Einblicke in die Entwicklungsbiologie und kann verwendet werden, um bessere Krankheitsmodelle für das Screening von Medikamenten und die Entwicklung von Therapien zu erstellen.
Muster-Einreichungsanforderungen
| Probenart | Erforderlicher Betrag | Zusätzliche Hinweise |
|---|---|---|
| Zellen | Protein ≥ 400.000, Transkriptionsfaktoren ≥ 1.000.000 | Die Zellviabilität nach dem Auftauen sollte ≥ 85 % betragen. |
| Tiergewebe | Transkriptionsfaktoren ≥ 200 mg, Histone ≥ 100 mg | Stellen Sie die Stabilität der Proben sicher, indem Sie 1-2 Backup-Proben vorbereiten. |
Demonstrationsergebnisse

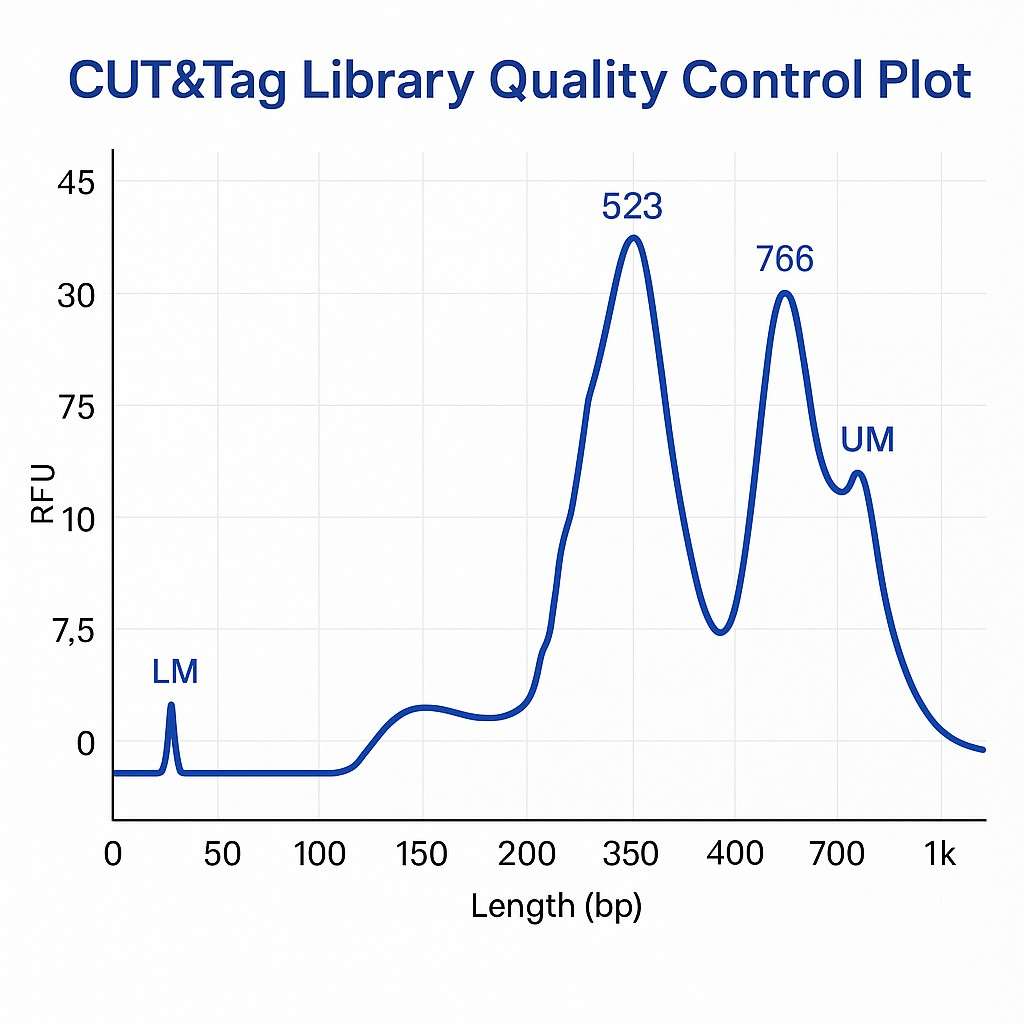
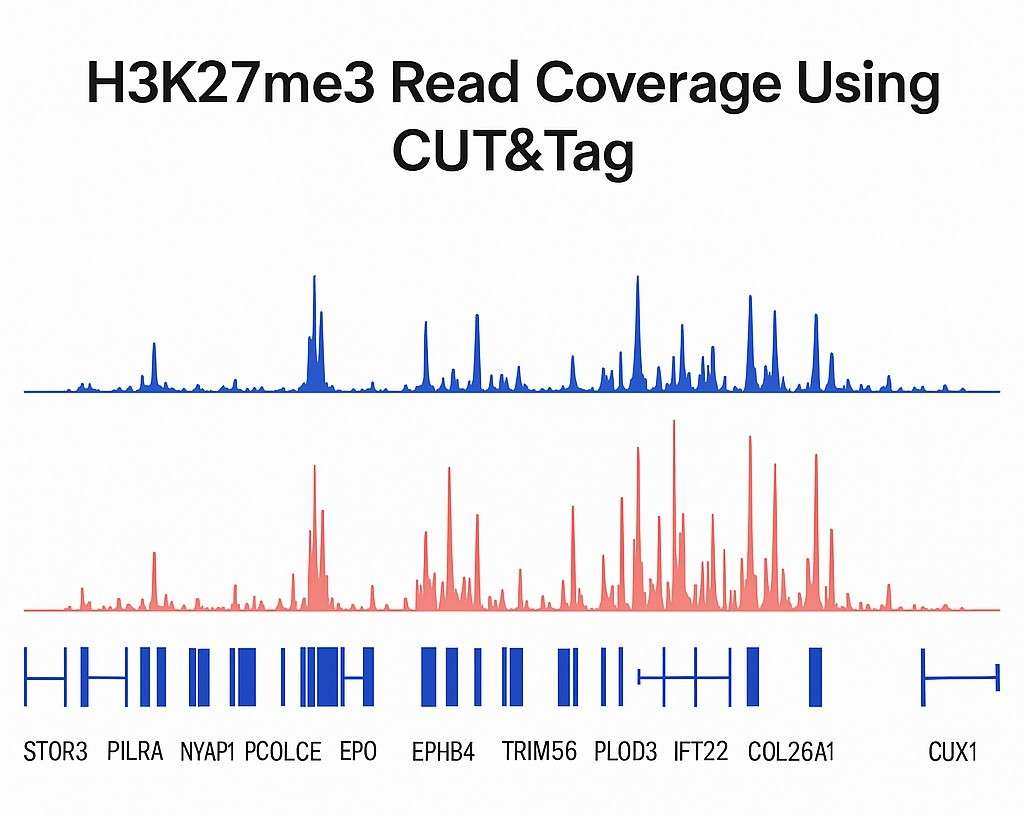
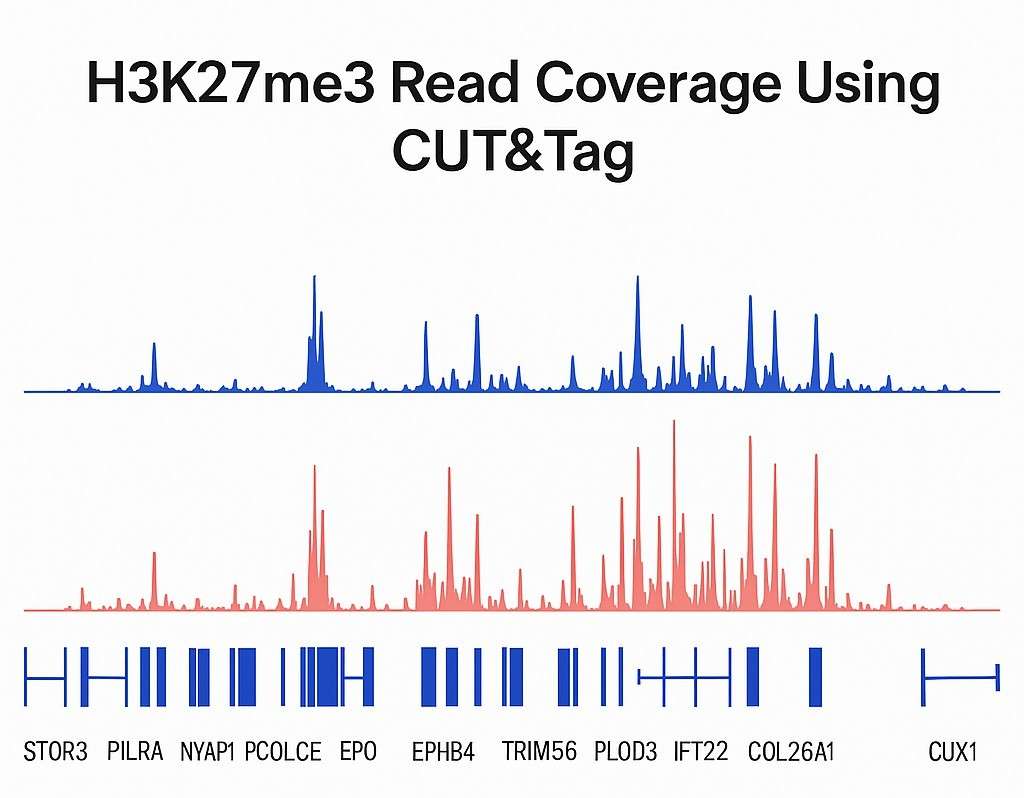
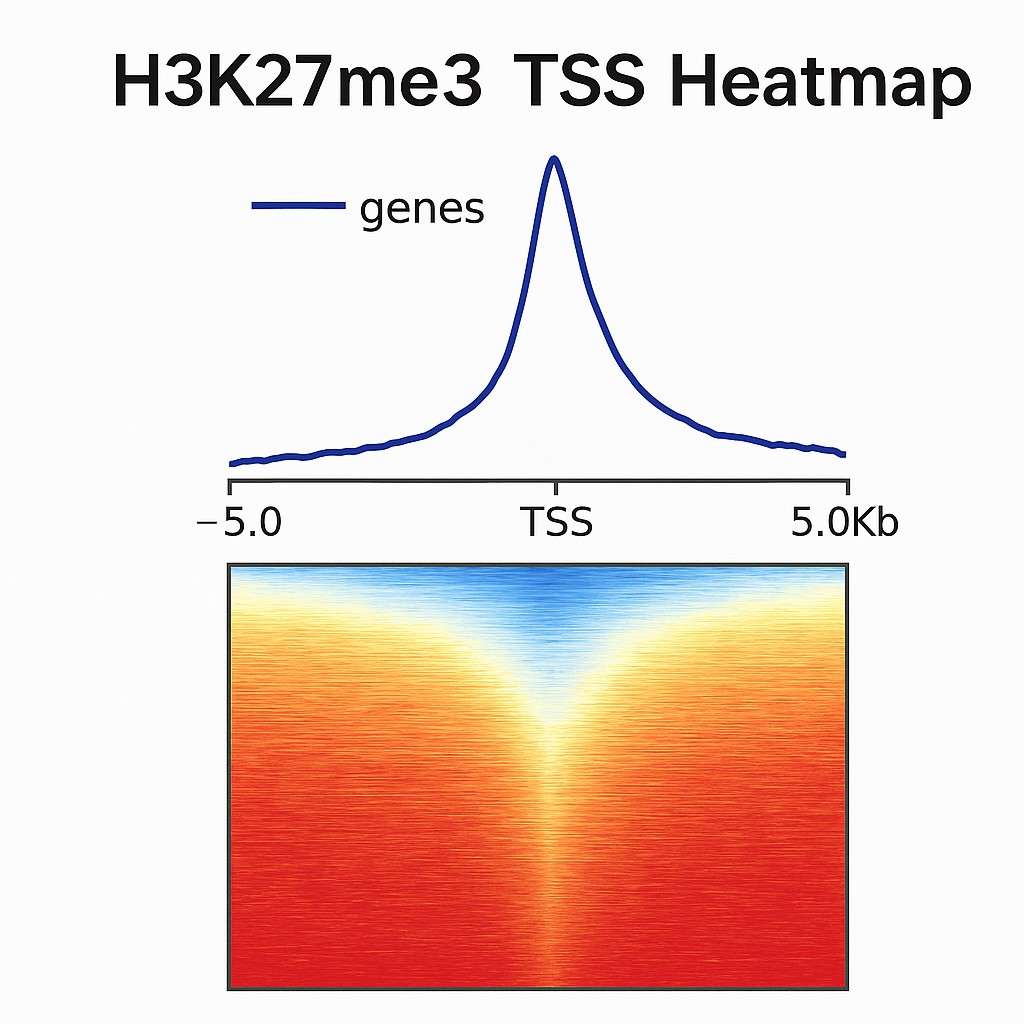
CUT&Tag Seq FAQs
1. Was ist die minimale Anzahl an Zellen, die für CUT&Tag-Sequenzierung erforderlich ist?
Einer der wichtigsten Vorteile von CUT&Tag-Sequenzierung ist die Fähigkeit, mit Proben mit niedrigem Input zu arbeiten. Typischerweise können Forscher zuverlässige Ergebnisse mit so wenigen wie 1.000 bis 10.000 ZellenDies macht die Technik äußerst geeignet für die Einzelzell-Epigenomik und Studien, die seltene oder schwer isolierbare Zellpopulationen betreffen.
2. Wie vergleicht sich CUT&Tag mit ATAC-seq und ChIP-seq?
CUT&Tag ist empfindlicher und benötigt im Vergleich zu traditionellen Methoden wie weniger Zellen. ChIP-seq, was es kostengünstiger und effizienter macht, um Protein-DNA-Interaktionen zu untersuchen. Während ChIP-seq benötigt große Stichprobengrößen und kann anfälliger für Hintergrundgeräusche sein, bietet CUT&Tag eine höhere Spezifität und erfordert weniger Ausgangsmaterial.
ATAC-seq ist eine Technik zur Kartierung der Chromatinzugänglichkeit und konzentriert sich auf die Identifizierung offener Regionen des Genoms. Während beide CUT&Tag und ATAC-seq geben Sie Einblicke in die Chromatinstruktur, CUT&Tag konzentriert sich stärker auf Protein-DNA-Interaktionen und kann hochauflösende Daten für das Studium von Histonmodifikationen und der Bindung von Transkriptionsfaktoren liefern. Für einen detaillierteren Vergleich erkunden Sie unseren Abschnitt zu ATAC-seq.
3. Was sind die Anforderungen an die Probenvorbereitung für CUT&Tag-Sequenzierung?
Die Probenvorbereitung für CUT&Tag-Sequenzierung ist unkompliziert, erfordert jedoch sorgfältige Aufmerksamkeit für Details. Typischerweise umfasst der Prozess:
- Zellisolierung: Zellen von Interesse werden aus Gewebeproben isoliert.
- Zellfixierung: Die Zellen werden fixiert, um Protein-DNA-Interaktionen zu erhalten.
- Antikörperinkubation: Ein spezifischer Antikörper für das interessierende Protein wird hinzugefügt, um an das Protein zu binden.
- Tagmentierung: Die DNA in der Nähe des an das Protein gebundenen Antikörpers wird zum Sequenzieren markiert.
Forscher sollten sicherstellen, dass die Probe von hoher Qualität ist, da eine schlechte Probenvorbereitung die Genauigkeit der Ergebnisse beeinträchtigen kann. Bei der Arbeit mit niedrigem Input oder seltenen Zellen können spezialisierte Protokolle erforderlich sein.
4. Wie lange dauert es, Ergebnisse von CUT&Tag-Sequenzierung zu erhalten?
Der Durchlaufzeit für CUT&Tag-Sequenzierung liegt typischerweise im Bereich von 3 bis 6 Wochen abhängig von Faktoren wie der Sequenzierungstiefe, der Komplexität der Probe und zusätzlichen bioinformatischen Analysen. Wenn jedoch eine beschleunigte Bearbeitung erforderlich ist, bieten viele Sequenzierungsanbieter schnellere Bearbeitungsoptionen gegen eine zusätzliche Gebühr an.
5. Kann CUT&Tag-Sequenzierung für die Einzelzellanalyse verwendet werden?
Ja, CUT&Tag-Sequenzierung ist besonders gut geeignet für Einzelzell-Epigenomik aufgrund seiner geringen Anforderungen an die Probenmenge. Dies ermöglicht es, einzelne Zellen zu untersuchen und die zelluläre Heterogenität zu erforschen, was entscheidend für das Verständnis komplexer biologischer Prozesse und Krankheiten ist.
6. Welche Art von bioinformatischer Analyse ist für CUT&Tag-Sequenzierungsdaten erforderlich?
Nach der Sequenzierung erfordert CUT&Tag-Daten eine bioinformatische Analyse, um die Ergebnisse zu interpretieren. Dies umfasst typischerweise:
- Leseausrichtung: Abbildung der Sequenzierungsreads auf ein Referenzgenom.
- Peak-Identifizierung: Bestimmung von Regionen des Genoms, die reich an Protein-DNA-Interaktionen sind.
- Differenzielle Analyse: Vergleich von Bindungsmustern unter verschiedenen Bedingungen oder Proben.
Je nach Komplexität des Projekts benötigen Forscher möglicherweise zusätzliche maßgeschneiderte Analysen, wie z. B. Motif-Entdeckung oder Integration mit anderen Omics-Daten. Viele Sequenzierungsanbieter bieten bioinformatische Dienstleistungen an, um diese Analysen zu unterstützen.
7. Ist CUT&Tag-Sequenzierung für Studien zu Histonmodifikationen geeignet?
Ja, CUT&Tag-Sequenzierung ist eine ausgezeichnete Methode zur Kartierung Histonmodifikationen über das gesamte Genom. Es bietet eine hohe Auflösung und Spezifität, die es Forschern ermöglicht, spezifische Histonmarkierungen zu identifizieren, die mit der Genaktivierung oder -repression verbunden sind. Dies macht es zu einem leistungsstarken Werkzeug für das Studium der Chromatinstruktur und der Genregulation.
Für zusätzliche Informationen oder wenn Sie weitere Fragen haben zu CUT&Tag-SequenzierungZögern Sie nicht, unsere umfassenden Ressourcen zu durchsuchen oder unser Team für persönliche Unterstützung zu kontaktieren.


