
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben

Was ist Amplicon-Sequenzierung?
Amplicon-Sequenzierung ist eine gezielte Next-Generation-Sequenzierung (NGS) Ansatz, der speziell entworfene Primer verwendet, um ausgewählte genomische Regionen mittels PCR zu amplifizieren, gefolgt von Hochdurchsatz-Sequenzierung der Amplifikate. Diese Technik ermöglicht die präzise Erkennung genetischer Varianten innerhalb definierter Loci und wird häufig in Studien zu Genmutationen, mikrobieller Vielfalt und Biomarker-Screening eingesetzt.
Wie es funktioniert
- Zielauswahl und Primerdesign – Primer werden entworfen, um spezifische genomische Regionen von Interesse basierend auf den Forschungszielen zu amplifizieren.
- PCR-Amplifikation – Die Zielregionen werden aus der Proben-DNA amplifiziert, um hochabundante Amplicons zu erzeugen.
- Bibliothekskonstruktion und Sequenzierung – Amplicons werden mit Adaptern ligiert und in Sequenzierungsbibliotheken vorbereitet, anschließend werden sie mit Plattformen wie Illumina oder PacBio sequenziert.
- Datenanalyse – Sequenzierungsdaten werden für die Ausrichtung, Variantenbestimmung, Sequenzassemblierung oder taxonomische Klassifikation verarbeitet.
Dieses Verfahren ist ideal für die Hochdurchsatzdetektion von Mutationen in mehreren Proben und ermöglicht die gleichzeitige Untersuchung von Hunderten bis Tausenden von Amplicon-Loci.

Warum Amplicon-Sequenzierung verwenden?
Die Amplicon-Sequenzierung ist ein hocheffizienter und kostengünstiger gezielter Sequenzierungsansatz, der sich ideal für die eingehende Analyse spezifischer genomischer Regionen, mikrobieller Gemeinschaften oder funktioneller Genelemente eignet. CD Genomics bietet umfassende, anpassbare Amplicon-Sequenzierungslösungen – unterstützt Ihre Forschung von der Primer-Design bis zur bioinformatischen Analyse.
- Hochgradig zielgerichtet & genau
Durch die Amplifikation nur der Regionen von Interesse mit spezifischen Primern gewährleistet diese Methode eine präzise Variantenerkennung mit hoher Sensitivität – ideal für locus-spezifische Validierungen und funktionale Studien. - Hoher Durchsatz & Multiplexing
In der Lage, Hunderte bis Tausende von Zielregionen pro Durchlauf zu analysieren, was es für großangelegte Screenings, die Profilierung mikrobieller Diversität und die parallele Probenverarbeitung geeignet macht. - Plattformflexibilität und breites Amplicon-Spektrum
Kompatibel mit sowohl Illumina (Short-Read) als auch PacBio (Long-Read) Plattformen. Unterstützt Amplicons von 100 bp bis zu 10 kb und erfüllt eine Vielzahl von Forschungsbedürfnissen. - Empfindlich gegenüber seltenen Varianten
Mit ultra-tiefer Sequenzierung ist es hervorragend darin, niedrigfrequente somatische Mutationen zu erkennen und komplexe mikrobielle Mischungen aufzulösen, sodass wichtige Erkenntnisse nicht übersehen werden.

Amplicon-Sequenzierung vs. Andere NGS-Methoden
| Merkmal | Amplicon-Sequenzierung | Gezielte Erfassungssequenzierung | Whole Genome Sequencing (WGS) - Gesamtes Genom-Sequenzierung (WGS) |
|---|---|---|---|
| Zielregion | Spezifische PCR-amplifizierte Loci | Breitere Regionen über hybride Sonden | Ganzes Genom |
| Sequenzierungstiefe | Ultra-tief (>1000×) | Mäßig bis tief (200–800×) | Niedrig bis moderat (~30×) |
| Datenvolumen / Kosten | Niedrig | Mittel | Hoch |
| Empfindlichkeit der Variantenerkennung | Hoch (ideal für seltene/niedrigfrequente Varianten) | Mittel | Niedrig bis mittel |
| Anwendungsfälle | 16S/18S/ITS , CRISPR-Validierung Antikörperrepertoire | Krebs-Panels, Gene seltener Krankheiten | Populationsgenetik strukturelle Varianten |
Die Wahl der richtigen Amplicon-Sequenzierungsplattform und Leselänge
Bei CD Genomics bieten wir drei Amplicon-Sequenzierungsoptionen an, die auf Ihre Amplicon-Länge, Forschungsziele und Datenanforderungen zugeschnitten sind:
| Sequenzierungstyp | Ampliconlänge | Plattform | Leseumfang | Typische Anwendungen |
|---|---|---|---|---|
| Standard-Amplikon-Sequenzierung | 100–250 bp | Illumina MiSeq | 2×150 bp | SNP-Genotypisierung , Bearbeitung der Standortvalidierung, schnelle Belastungsprüfung |
| Mittel-lange Amplicon-Sequenzierung | 250–550 bp | Illumina MiSeq / NextSeq | 2×250 bp oder 2×300 bp | Hochvariablen Regionen (z. B. 16S V3-V4), Antikörper schwere/leichte Ketten |
| Lange Amplicon-Sequenzierung | >550 bp bis zu ~10 kb | PacBio Sequel | HiFi CCS (hochauflösende Langlesungen) | Vollständig 16S/ITS , gepaarte Antikörperketten, Phasierung von Varianten |
Plattform-Highlights:
- Illumina-Plattform: Bietet hohe Sequenziergenauigkeit; ideal für kurze bis mittellange Amplicons. Unterstützt eine hohe Multiplexkapazität, was sie gut geeignet für Hochdurchsatzstudien macht.
- PacBio PlattformErmöglicht Langzeit-, hochauflösende Sequenzierung. Am besten geeignet für strukturell komplexe oder lange Amplicons, die eine Phasierung oder vollständige Analyse erfordern.
Empfehlungen:
- Für Amplicons <250 bp empfehlen wir die Illumina-Paarendsequenzierung 2×150 bp.
- Für Amplicons zwischen 250–550 bp empfehlen wir Illumina 2×250 bp oder 2×300 bp Konfigurationen.
- Für Amplicons >550 bp oder Projekte, die vollständige Sequenzen erfordern, empfehlen wir die PacBio-Plattform mit HiFi (High-Fidelity) Reads für eine einzelmolekulare genaue Sequenzierung.
Unser Amplicon-Sequenzierungsprozess: Von der Beratung bis zur Berichterstattung
Unser modularer Workflow gewährleistet eine standardisierte Qualitätskontrolle in jedem Schritt und ermöglicht flexible Anpassungen basierend auf den Projektanforderungen:

Ziele definieren
Wählen Sie die Sequenzierungsplattform und -tiefe aus.
Workflow bestätigen

Musteranmeldung
DNA/PCR-Qualitätskontrolle
Optionale PCR-Amplifikationsdienstleistung

Adapter-Ligation
Indizierung und Pooling
Bibliotheksqualitätsprüfung

Illumina- oder PacBio-Plattformen
Kurze oder lange Texte
Anpassbare Sequenzierungstiefe

Datenqualitätskontrolle
Variantenerkennung und taxonomische Analyse
Lieferung des Abschlussberichts
Forschungsanwendungen der Amplicon-Sequenzierung
Unsere Amplicon-Sequenzierungsdienste werden in verschiedenen Forschungsbereichen breit angewendet und ermöglichen eine präzise und effiziente Analyse genomischer Variationen und komplexer Sequenzinformationen. Zu den wichtigsten Anwendungsbereichen gehören:
- Erkennung genetischer Varianten
Genaues Identifizieren von SNPs, somatischen Mutationen und komplexen genetischen Varianten zur Unterstützung verschiedener genetischer Studien und Genotypisierungen. - Mikrobiomforschung
Sequenzierung von 16S rRNA, 18S rRNA und ITS-Regionen zur Analyse der mikrobiellen Diversität, phylogenetischen Profilierung und ökologischen Studien. - Immunsystem-Repertoire-Sequenzierung
Gezielte Sequenzierung von Antikörper-Schwer- und Leichtketten zur Unterstützung der Immunvielfalt-Profilierung und der Entwicklung therapeutischer Antikörper. - Validierung der Genbearbeitung
Bewertung der Effizienz der CRISPR/Cas9-Bearbeitung und der Off-Target-Effekte, um Genauigkeit und Zuverlässigkeit in Genom-Editierungsexperimenten sicherzustellen. - Funktionales Gen-Screening
Hochdurchsatz-Screening spezifischer Genregionen zur Unterstützung von Genfunktionsstudien und der Entdeckung neuer Ziele. - Plasmidbibliotheksanalyse
Umfassende Analyse von Plasmidbibliotheken hinsichtlich Vielfalt und struktureller Merkmale zur Unterstützung von molekularer Klonierung und Gentechnik.

Amplicon-Sequenzierung Bioinformatik- und Datenanalyse-Dienste
Wir bieten umfassende und anpassbare Lösungen an. Bioinformatik Lösungen für Amplicon-Sequenzierungsprojekte, die sowohl Kurz- als auch Langlese-Sequenzierungsplattformen unterstützen. Unsere Dienstleistungen helfen Kunden, den vollen Wert ihrer Daten zu erschließen und eine genaue Variantenerkennung sowie funktionale Interpretation zu erreichen.
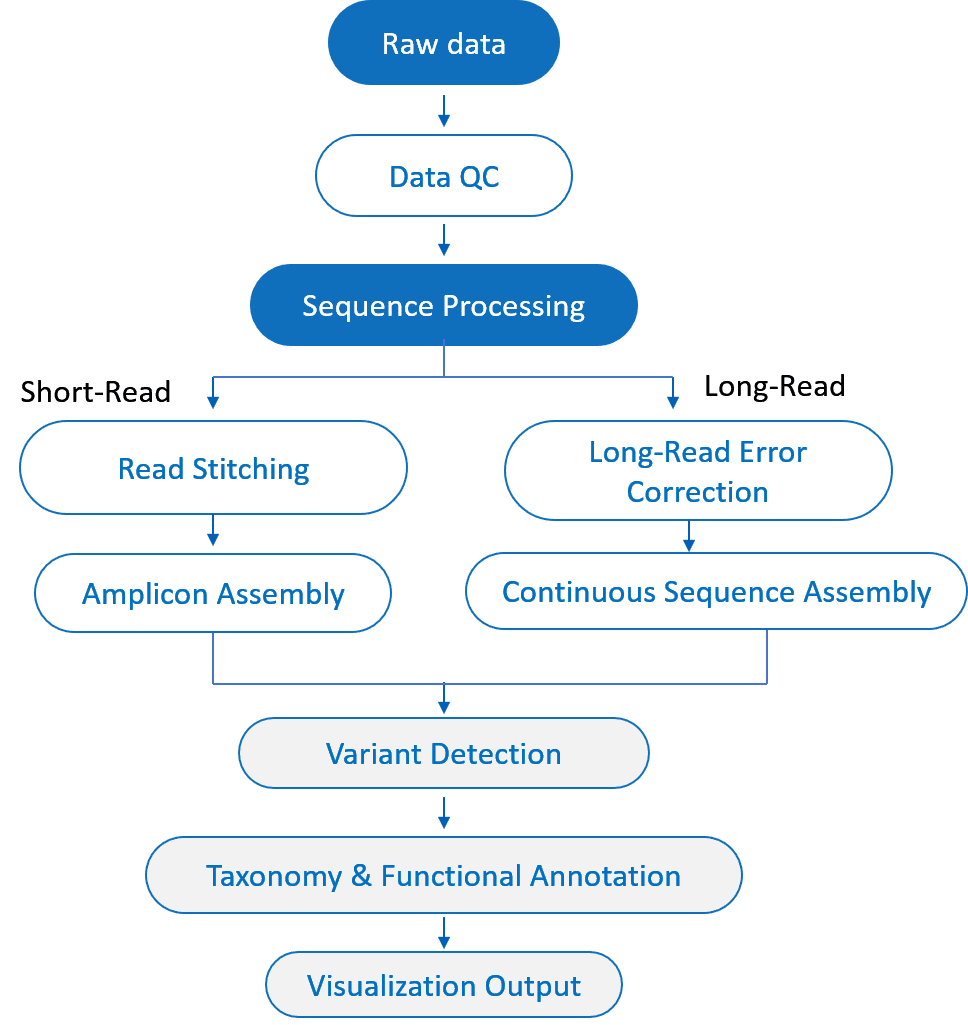
Short-Read Amplicon-Sequenzierung (Illumina) Analyse
- Datenqualitätskontrolle und Vorverarbeitung
Entfernung von Adaptersequenzen und niedrigqualitativen Reads, Zusammenführung von Paar-End-Reads zur Sicherstellung hochwertiger Daten. - Rauschunterdrückung & Variantenentdeckung
Fortgeschrittene Algorithmen (z. B. DADA2, UNOISE) zur Geräuschentfernung und Chimärenfilterung; genaue Identifizierung von SNPs und Indels. - Taxonomische Annotation und Klassifikation
Hocheffiziente Artenannotation basierend auf 16S, 18S, ITS und anderen kuratierten Datenbanken. - Vielfaltsanalyse
Alpha- und Beta-Diversitätsbewertungen zur Unterstützung des Vergleichs der mikrobiellen Gemeinschaftsstruktur und ökologischer Statistiken. - Funktionale Vorhersage und Pfadanalyse
Systematische Vorhersage der Genfunktion zur Aufdeckung potenzieller biologischer Funktionen und Stoffwechselwege. - Datenvisualisierung & Berichterstattung
Reiche grafische Ausgaben und intuitive Berichte zur Förderung eines tiefen Verständnisses der Sequenzierungsergebnisse.
Long-Read-Amplicon-Sequenzierung (PacBio / ONT) Analyse
- Hochgenaue Korrektur von Langlesen
CCS (Circular Consensus Sequencing) und Fehlerkorrekturalgorithmen verbessern die Lesegenauigkeit und Datenzuverlässigkeit. - Vollständige Amplicon-Assemblierung
Erhalten Sie vollständige Amplicon-Sequenzen ohne die Notwendigkeit des Zusammenfügens, was eine genaue Erkennung komplexer und langreichweitiger Varianten ermöglicht. - Phasierung und Erkennung struktureller Varianten
Erkennen Sie SNPs, Indels und strukturelle Varianten aus vollständigen Lesevorgängen, die eine hochauflösende Variantenphasenunterstützung bieten. - Hochauflösende taxonomische und funktionale Annotation
Arten- und Genfunktionsannotation in höherer Auflösung basierend auf vollständigen Sequenzalignments. - Fortgeschrittene Profilierung mikrobieller Gemeinschaften
Nutzen Sie vollständige Lesevorgänge für eine umfassende Analyse der mikrobiellen Gemeinschafts- und Funktionsvielfalt. - Benutzerdefinierte Berichterstattung
Die Liefergegenstände umfassen Karten zu strukturellen Variationen, vollständige Details zu Varianten und eine umfassende funktionale Interpretation zur Unterstützung wissenschaftlicher Publikationen.
Beispielanforderungen und Qualitätsrichtlinien für Amplicon-Sequenzierung
Um hochwertige Sequenzierungsergebnisse zu gewährleisten, bietet CD Genomics klare Richtlinien für Probenarten und Eingabebedürfnisse. Im Folgenden finden Sie eine schnelle Übersicht über empfohlene Mengen und Qualitätskriterien:
| Probenart | Empfohlene Eingabe | Reinheit (OD260/280) | Anforderungen | Notizen |
|---|---|---|---|---|
| Reinige PCR-Produkte | ≥1 µg (min. 500 ng) | 1,8–2,0 | ≥20 ng/μL; hochqualitativ; Größe entspricht den Plattform-Spezifikationen | Einzelnes, spezifisches Band; keine unspezifischen Produkte |
| Unreinigte PCR-Produkte | Variable | — | Akzeptabel, aber eine Reinigung wird dringend empfohlen, um eine optimale Sequenzierungsqualität zu gewährleisten. | — |
| Fragmentierte DNA | Ausreichende Menge | — | Erfordert einheitliche Größe und Kompatibilität mit der Zielregion. | Für spezifische Amplicon-Strategien |
| Genomisches DNA (gDNA) | ≥500 ng | 1,8–2,0 | Hohe Reinheit; ≥20 ng/μL; keine Zersetzung | Ideal für PCR-basierte Amplifikation |
| Restriktionsenzym-Schnitte | Angemessene Menge | — | Vollständige Verdauung; frei von Hemmstoffen | Geeignet für die nachgelagerte Bibliotheksvorbereitung |
| Plasmide | Angemessene Menge | — | Muss gereinigt werden; die Integrität des Zielinserts sicherstellen. | — |
💡 Hinweis: Dies sind allgemeine Empfehlungen. Bei projektspezifischen Bedürfnissen wenden Sie sich bitte an unser technisches Team für maßgeschneiderte Unterstützung.
Warum CD Genomics für Amplicon-Sequenzierung wählen?
CD Genomics ist auf die Bereitstellung von hochwertigen, hochdurchsatzfähigen Amplicon-Sequenzierungsdiensten spezialisiert und nutzt fortschrittliche Illumina- und PacBio-Plattformen, um unterschiedlichen Anforderungen an Ampliconlängen und -komplexität gerecht zu werden. Wir setzen uns dafür ein, genaue und zuverlässige Daten zur Variantenanalyse bereitzustellen, die genomische Forschung, Mikrobiomstudien und funktionale Gen-Screenings in verschiedenen Bereichen unterstützen.
- Multiplattform-Technologieunterstützung
Umfassende Abdeckung von Hunderten bis Zehntausenden von Basenpaaren mithilfe von Illumina-Kurzlesungen und PacBio-Langlesungen, die verschiedene experimentelle Designs berücksichtigen. - Überlegene Datenqualität und Genauigkeit
Strenge Qualitätskontrollprozesse gewährleisten eine hohe Abdeckung und eine tiefe Sequenzierungstiefe für die präzise Erkennung von Varianten mit niedriger Frequenz. - Angepasste bioinformatische Analyse
Professionelle Sequenzassemblierung, Variantenerkennung und funktionale Annotation mit intuitiven Visualisierungsberichten, um Kunden zu helfen, wertvolle Erkenntnisse schnell zu gewinnen. - End-to-End Professionelle Unterstützung und Schnelle Reaktion
Erfahrene Teamleitung während des Projektentwurfs, der Qualitätskontrolle von Proben, der Sequenzierung und der Datenlieferung, um eine effiziente und reibungslose Projektabwicklung sicherzustellen.

Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:
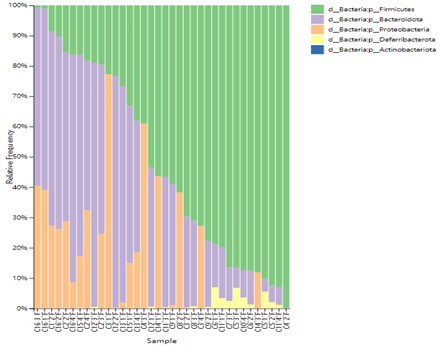
Die Taxonomieverteilung aller Proben auf der Ebene der Stammklassifikation.
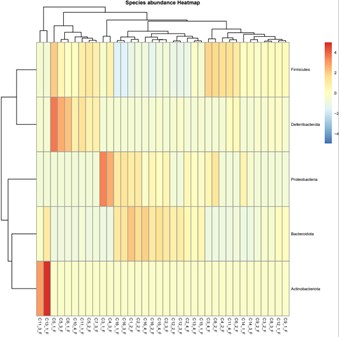
Artenhäufigkeit Wärmebild.
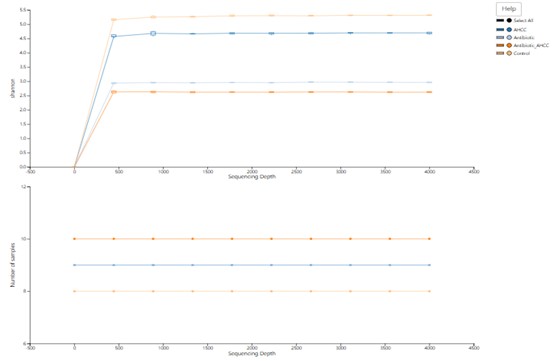
Seltenheitskurve der sequenzierten Reads für Proben (Die obige Abbildung) & Die Sequenzierungstiefe der Proben (Die untere Abbildung).
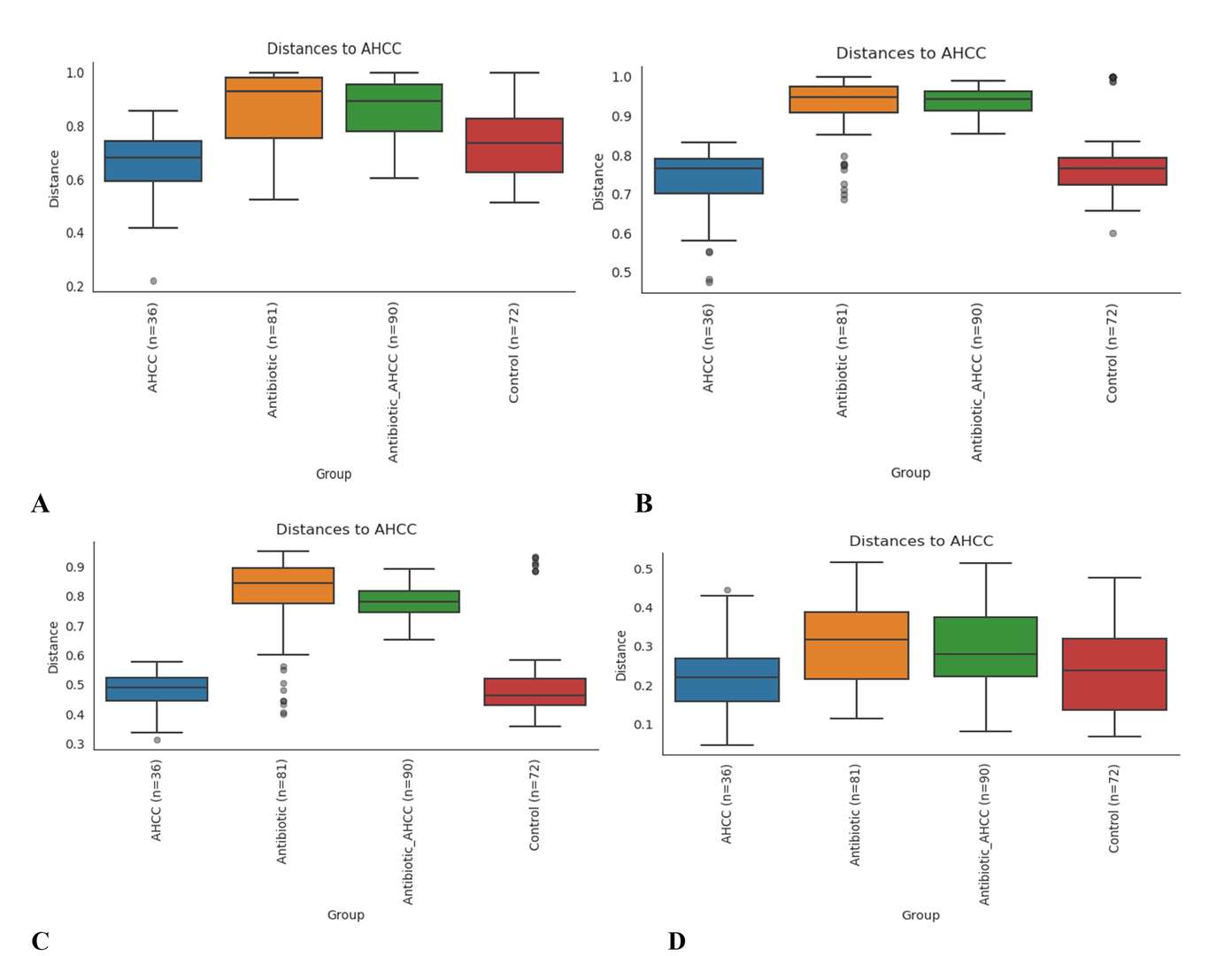
Boxplot-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichteten Unifrac (C) und gewichteten Unifrac (D).
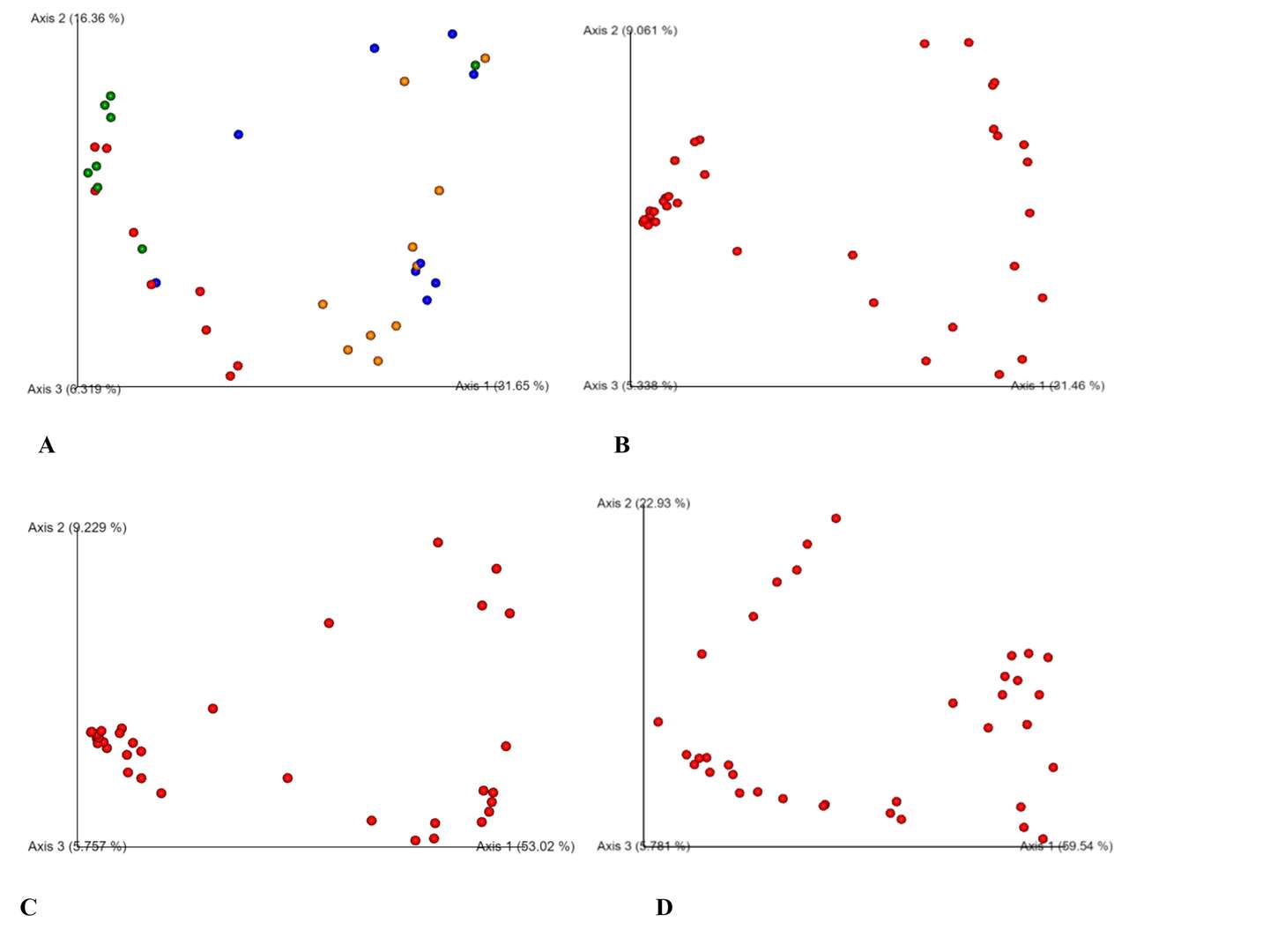
PCoA-Analyse basierend auf Bray-Curtis (A), binärem Jaccard (B), ungewichtetem Unifrac (C) und gewichtetem Unifrac (D).
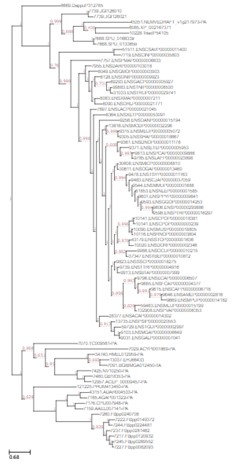
UPGMA-Clusterbaum.
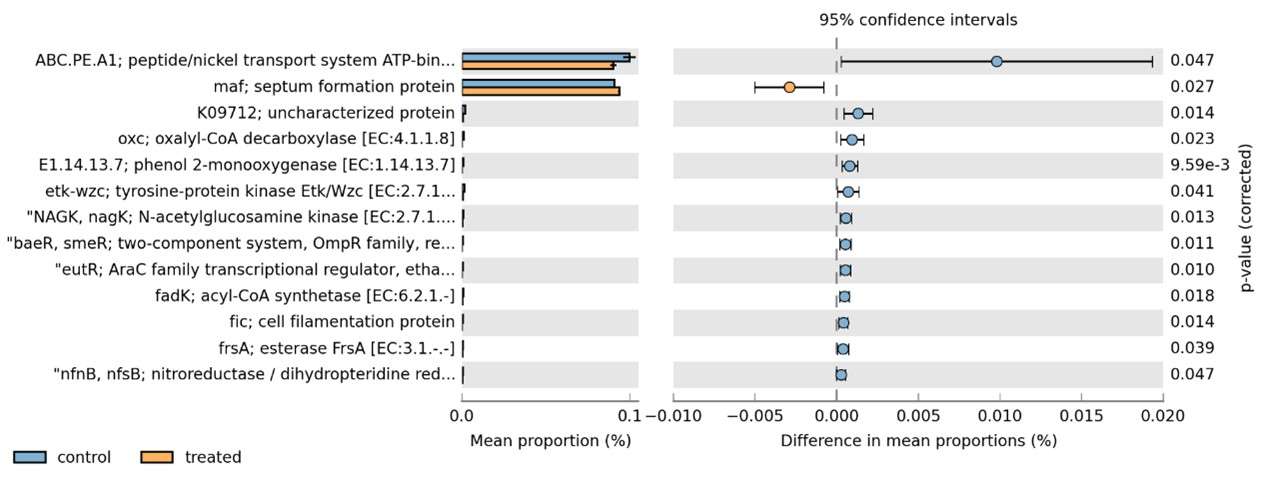
Mittelwert der behandelten und Kontrollgruppe.
Kladogram.

LDA-PUNKTZAHL.
Amplicon-Sequenzierungs-FAQs
1. Was ist der Unterschied zwischen gezielter Sequenzierung und Amplicon-Sequenzierung?
Amplicon-Sequenzierung umfasst die PCR-Amplifikation spezifischer genomischer Regionen, gefolgt von der Sequenzierung, was hohe Spezifität und Zielgenauigkeit aufgrund des präzisen Designs der Primer gewährleistet. Sie ist besonders geeignet für die Analyse kleiner, definierter Regionen des Genoms, wie beispielsweise bei der Analyse genetischer Variationen und der mikrobiellen Profilierung. Im Gegensatz dazu, gezielte Sequenzierung umfasst Methoden wie Hybrid-Capture und Sonden-basierte Anreicherung, um gezielt größere genomische Regionen oder mehrere Gene ohne vorherige Amplifikation zu sequenzieren. Dies ermöglicht eine umfassendere Analyse der ausgewählten Bereiche, kann jedoch je nach Effizienz des Anreicherungsprozesses variable On-Target-Raten aufweisen. Die Amplicon-Sequenzierung erreicht aufgrund ihrer Natur im Gegensatz zu anderen Methoden überlegene On-Target-Raten. gezielte Sequenzierung Methoden, die diese Effizienz dem präzisen Design von Primern zuschreiben. Dieser Ansatz findet besondere Anwendung in Aufgaben wie Genotypisierung durch Sequenzierung sowie die Erkennung von Keimbahn-einzelnen Nukleotid-Polymorphismen (SNPs), Insertionen und Deletionen (Indels) sowie bekannten genetischen Fusionen.
2. Was sind die Hauptanwendungen der Amplicon-Sequenzierung?
Die Amplicon-Sequenzierung dient als ein entscheidendes Werkzeug in verschiedenen wissenschaftlichen Bereichen, einschließlich, aber nicht beschränkt auf die folgenden Anwendungen:
- Genetische Variationsanalyse: Aufdeckung von Einzel-Nukleotid-Polymorphismen (SNPs), Insertionen, Deletionen und anderen erblichen genetischen Modifikationen.
- Mikrobiom-Untersuchungen: Profilierung mikrobieller Gemeinschaften durch Sequenzierung von Marker-Genen wie der 16S ribosomalen RNA.
- Onkologische Untersuchung: Erkennung somatischer Mutationen und genetischer Veränderungen in Tumorproben.
- Forschung zu Erbkrankheiten: Untersuchung der genetischen Grundlagen von erblichen Störungen.
- Umwelterhebungen: Bewertung der Biodiversität und Identifizierung spezifischer Organismen innerhalb von Umweltproben.
3. Was ist der Unterschied zwischen Amplicon-Sequenzierung und Whole-Genome-Sequenzierung (WGS)?
Die Amplicon-Sequenzierung zielt auf spezifische genomische Regionen ab, indem sie diese mit PCR amplifiziert, bevor sie sequenziert werden. Dies ermöglicht eine hohe Spezifität und Tiefe bei der Analyse kleiner, definierter Regionen, wie zum Beispiel bei der Erkennung von Mutationen oder der Profilierung mikrobieller Gemeinschaften. Im Gegensatz dazu, Whole-Genome-Sequenzierung (WGS) Sequenzen das gesamte Genom ohne vorherige Selektion oder Amplifikation und bietet einen umfassenden Überblick über alle genetischen Informationen, was ideal ist, um neuartige Varianten zu entdecken und ein vollständiges genetisches Profil zu erhalten, jedoch ressourcenintensiver ist und weniger auf spezifische Interessensgebiete fokussiert ist.
4. Wie wählen Sie die Zielregionen für die Amplicon-Sequenzierung aus?
Die Auswahl der Zielsegmente in der Amplicon-Sequenzierung hängt von den Zielen der Studie und der biologischen Bedeutung dieser Segmente ab. Wichtige Faktoren sind Assoziationen mit Krankheiten, genetische Marker, Regionen mit bemerkenswerter Variabilität und funktionale Relevanz. Die Zusammenarbeit mit Bioinformatikern und die Nutzung von Datenbanken wie dbSNP und ClinVar können die präzise Identifizierung der Zielregionen erleichtern.
5. Welche Arten von bioinformatischen Analysen können mit Amplicon-Sequenzierungsdaten durchgeführt werden?
- Variantenerkennung: Erkennung von SNPs, Insertionen, Deletionen und verschiedenen genetischen Variationen.
- Analyse der mikrobiellen Vielfalt: Bewertung der Zusammensetzung und Häufigkeit mikrobieller Konsortien.
- Phylogenetische Untersuchung: Erforschung der evolutionären Verbindungen zwischen Sequenzen.
- Funktionale Erläuterung: Assoziation genetischer Variationen mit plausiblen funktionalen Implikationen.
6. Kann die Amplicon-Sequenzierung seltene Varianten nachweisen?
Ohne Zweifel zeigt die Amplicon-Sequenzierung eine bemerkenswerte Sensitivität, die die Erkennung seltener Varianten bei niedrigen Vorkommen ermöglicht. Dieses Merkmal macht sie anwendbar für Situationen wie das Auffinden von Mutationen bei Krebs und die Bewertung der mikrobiellen Vielfalt.
7. Wie stellt CD Genomics den Sequenzierungserfolg in Regionen mit hohem GC-Gehalt sicher?
Wir verwenden eine 3-Schichten-Strategie, um Ertrag und Genauigkeit zu maximieren:
- Optimierte Bibliotheksvorbereitung:
Methylierungsadaptive Polymerasen reduzieren die GC-Bias während der Amplifikation. - Verbesserte QC-Überwachung:
Echtzeit-Nanoporen-Sensorik gewährleistet intakte, hochwertige Fragmente. - Bioinformatik-Korrektur:
Fortgeschrittene Algorithmen kompensieren die GC-skeptische Häufigkeit in nachgelagerten Daten.
Amplicon-Sequenzierungs-Fallstudien
Kundenveröffentlichungshighlight
Mikrobielle Anpassung und Reaktion auf hohe Ammoniakkonzentrationen und Niederschläge während der anaeroben Vergärung unter psychrophilen und mesophilen Bedingungen
Journal: Wasserforschung
Veröffentlicht: 1. Oktober 2021
Hintergrund
Hohe Ammoniakkonzentrationen (TAN >1,5 g/L) sind eine Hauptursache für die Hemmung von Methan in der anaeroben Vergärung (AD), insbesondere unter mesophilen (37°C) und psychrophilen (22,6°C) Bedingungen. Phosphatniederschläge (z. B. Struvit) verschärfen den Systemzusammenbruch weiter und reduzieren den Methanertrag um mehr als 50 %. Diese Studie ist wegweisend für die Erforschung der mikrobiellen Ammoniak-Anpassungsmechanismen in psychrophilen Reaktoren und analysiert die langfristigen Auswirkungen von Niederschlägen auf methanogene Gemeinschaften.
Projektziele
- Mikrobielle AnpassungEntdecken Sie mikrobiologische Reaktionen auf hohen TAN (4.000 mg/L) in psychrophilen vs. mesophilen AD.
- Schlüssel-Konsortium-IdentifikationIdentifizieren Sie ammoniak-tolerante Methanogene und präzipitat-sensitive Taxa.
- Funktionale DynamikVerknüpfen Sie metagenomische Veränderungen mit Methanmetabolismuswegen.
CD Genomics Dienstleistungen
Als der zentrale Genomik-Partner lieferte CD Genomics:
- 16S rRNA-Amplikon-Sequenzierung
Plattform: Illumina MiSeq PE300
Zielregion: V4-V5 hypervariabel (optimiert für die Detektion von Archaeen).
Primers: 515F (5′-GTGYCAGCMGCCGCGGTAA) / 926R (5′-CCGYCAATTYMTTTRAGTTT).
Datenmenge: ~30.000 Reads/Stichprobe (alle Phasen der TAN-Anpassung abdeckend). - Ganzgenom Metagenomische Sequenzierung
Plattform: Illumina NovaSeq PE150.
Tiefe: 6 GB Rohdaten/probe (anfängliche vs. Endpunktproben).
DNA-Standards: Konzentration ≥50 ng/μL, 260/280-Verhältnis <1,8. - Bioinformatik-Analyse
Pipeline: MG-RAST v4.0.3.
Taxonomie: SILVA (16S-Klassifikation), GenBank (Metagenomklassifikation).
Funktionale Annotation: KEGG/Subsysteme-Datenbanken (e-Wert ≤10⁻²⁵, 90% Identität).
Vielfaltsmetriken: Shannon-Index, PCoA (Bray-Curtis-Distanz).
Wichtigste Erkenntnisse
- Ammoniak-Anpassung in einem psychrophilen Reaktor
- Dominanter Methanogen-Verschiebung:
- Bei TAN >3 g/L erreichte Methanocorpusculum eine relative Häufigkeit von 71 % (Metagenomdaten) und wurde zum dominierenden hydrogenotrophen Methanogen (Abb. 5a).
- Der bakterielle Konsortium verschob sich zu Enterococcaceae (Firmicutes) und stieg von 0,1% auf 80% Häufigkeit an (Abb. 4a).
- Funktionale Resilienz:
- Die KEGG-Analyse zeigte eine verstärkte Methanmetabolismus-Genaktivität (z.B. Umwandlung von Methylverbindungen) unter hohen TAN-Werten (Abb. 8).
- Dominanter Methanogen-Verschiebung:
- Niederschlagsinduzierter Zusammenbruch in mesophilen Reaktoren
- Methanerausbeute: Nach der Bildung von Niederschlägen um mehr als 50 % gesunken, ohne innerhalb von 50 Tagen eine Erholung.p<0,05).
- Methanogen-Depletion:
- Annotierte Methanogen-Gene wurden von über 300.000 auf weniger als 2.500 Treffer reduziert (Metagenomdaten, Abb. 5b).
- Artenersatz: Methanosarcina barkeri vertrieben M. mazei als das dominante ammoniak-tolerante Archaeon (Abb. 4b).
- α Diversität als Stabilitätsindikator
- Psychrophiler Reaktor: Die Diversität nahm nach erfolgreicher Ammoniak-Anpassung um 40% (Shannon-Index) ab (Abb. 3a).
- Mesophiler Reaktor: Stabile Vielfalt maskierte funktionale Ausfälle, belegt durch den anhaltenden Rückgang von Methan (Abb. 3b).
Zitierte Abbildungen
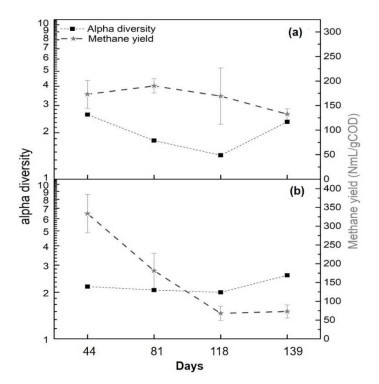 Abbildung 3. Alpha-Diversität und Methanertrag in experimentellen Reaktoren bei unterschiedlichen Ammoniakkonzentrationen (a) psychrophiler Reaktor (R1-CO) und (b) mesophiler Reaktor (R2-WW).
Abbildung 3. Alpha-Diversität und Methanertrag in experimentellen Reaktoren bei unterschiedlichen Ammoniakkonzentrationen (a) psychrophiler Reaktor (R1-CO) und (b) mesophiler Reaktor (R2-WW).
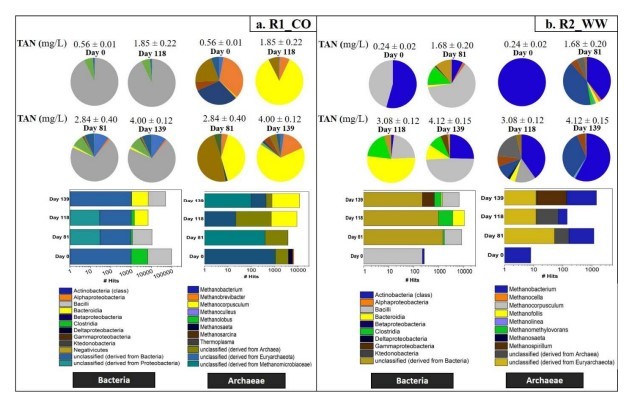 Abbildung 4. Profile von Bakterien und Archaeen aus 16S rRNA-Amplicon-Daten zusammen mit dem schrittweisen Anstieg der Ammoniakwerte.
Abbildung 4. Profile von Bakterien und Archaeen aus 16S rRNA-Amplicon-Daten zusammen mit dem schrittweisen Anstieg der Ammoniakwerte.
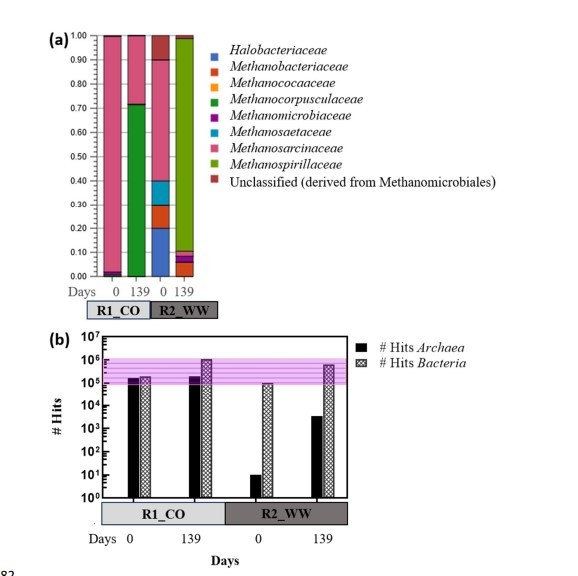 Abbildung 5. (a) Relative Häufigkeit des Archaea-Domains und (b) Anzahl der Treffer für Bakterien und Archaea-Domain in psychrophilen (R1-CO) und mesophilen (R2-WW) AD.
Abbildung 5. (a) Relative Häufigkeit des Archaea-Domains und (b) Anzahl der Treffer für Bakterien und Archaea-Domain in psychrophilen (R1-CO) und mesophilen (R2-WW) AD.
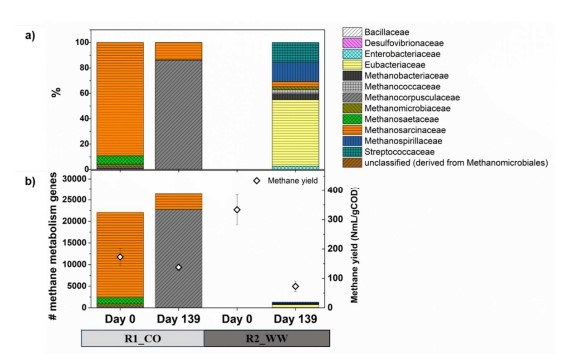 Abbildung 8. Anzahl der Genkopien für Methanmetabolismus (Anabolismus und Katabolismus) unter Verwendung der KEGG-Datenbank.
Abbildung 8. Anzahl der Genkopien für Methanmetabolismus (Anabolismus und Katabolismus) unter Verwendung der KEGG-Datenbank.
Implikationen
- ProzessoptimierungBereichernd Methanocorpusculum In der psychrophilen anaeroben Vergärung wird die Ammoniakverträglichkeit erhöht, wodurch der Heizenergiebedarf gesenkt wird.
- RisikominderungStruvitniederschlag verursacht irreversible Hemmung der Methanogenese, was eine Echtzeitüberwachung von Phosphat/pH erforderlich macht.
- Biotechnologische Ressource: M. barkeri und Methanocorpusculum dienen als Kandidaten für die Entwicklung von ammoniakresistentem Inokulum.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Unterschiedliche Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die therapieinduzierte Seneszenz.
Zeitschrift: Zellsterben & Krankheit
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generation eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginate
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genom-Analyse und Replikationsstudien des afrikanischen Grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


