
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben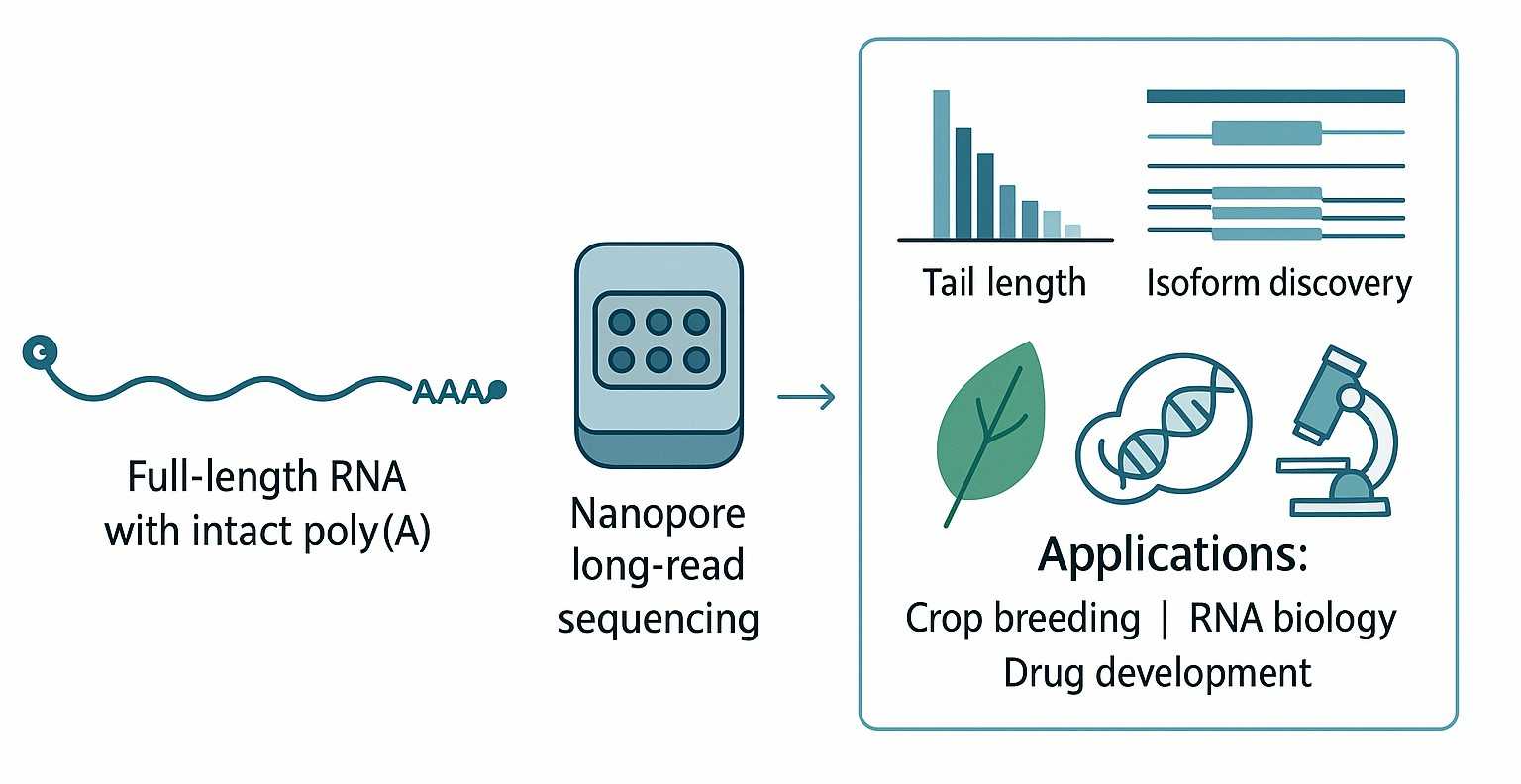
Warum Poly(A) und TAIL Iso-seq wichtig sind
Poly(A)-Schwänze sind mehr als einfache RNA-Erweiterungen. Sie fungieren als wichtige Regulatoren der Lebensdauer, Stabilität und Translation von mRNA. Standard-RNA-Seq-Methoden blenden diese Informationen oft aus oder übersehen sie und verpassen wertvolle regulatorische Signale.
TAIL Iso-seq schließt diese Lücke. Es erfasst vollständige cDNA, einschließlich intakter poly(A)-Schwänze, und bietet einen direkten Blick auf die Länge der poly(A)-Schwänze, deren Heterogenität und assoziierte APA-Ereignisse. Forscher können Veränderungen im 3'UTR mit miRNA-Bindung, identifizieren isoform-spezifische APA-Nutzungund untersuchen, wie die Schwanzlänge die Genexpression beeinflusst.
Dies macht TAIL Iso-seq zu einem wertvollen Werkzeug für:
- Untersuchung der RNA-Regulation in der Entwicklung
- Studie der Stressreaktionen bei Pflanzen
- Untersuchung der RNA-Stabilität in Krebsmodellen
- Unterstützung der Entwicklung von RNA-Therapeutika und Impfstoffen
Vorteile von TAIL Iso-seq mit CD Genomics
Vollständige Präzision
TAIL Iso-seq erfasst das vollständige Transkript, einschließlich 5'UTR, kodierender Region, 3'UTR und Poly(A)-Schwanz. Dies beseitigt Zusammenbaufehler, die bei RNA-seq mit kurzen Reads häufig auftreten, und bietet eine Auflösung auf Isoform-Ebene.
Umfassende Poly(A)-Profilierung
Unser Service quantifiziert die Verteilungen der Poly(A)-Schwanzlängen im Transkriptom und erkennt nicht-A-Reste (U, G, C), die die RNA-Stabilität und die Übersetzungseffizienz beeinflussen. Dies ermöglicht es Forschern, RNA-Dynamiken mit unübertroffener Detailgenauigkeit zu untersuchen.
APA- und 3'UTR-Analyse
TAIL Iso-seq identifiziert alternative Polyadenylierungsstellen (APA) und charakterisiert Veränderungen der 3'UTR-Längen. Dies hilft, APA mit Veränderungen in der miRNA-Bindung, RNA-Lokalisierung und posttranskriptioneller Regulation zu verknüpfen.
Umfangreiche bioinformatische Unterstützung
Wir liefern veröffentlichungsbereite Berichte mit Verteilungen der Schwanzlängen, APA-Karten, Isoformkatalogen und Analysen zur Anreicherung von Signalwegen. Visuelle Ausgaben erleichtern die Interpretation und können in Multi-Omics-Projekte integriert werden.
Technologischer Arbeitsablauf
CD Genomics bietet einen vollständigen TAIL Iso-seq-Workflow, von der RNA-Extraktion bis zur bioinformatischen Auswertung. Jeder Schritt ist auf Genauigkeit und Reproduzierbarkeit optimiert, um zuverlässige Erkenntnisse über die Poly(A)-Regulation und die Isoformvielfalt zu gewährleisten.
RNA-QualitätskontrolleDie Eingangs-RNA wird auf Integrität (RIN ≥7) und Reinheit überprüft.
Vollständige cDNA-ErfassungProtokolle bewahren intakte Poly(A)-Schwänze, und Barcodes können für multiplexe Projekte hinzugefügt werden.
Nanopore-Langzeit-SequenzierungLiest vollständige Transkripte, einschließlich 5'UTR, CDS, 3'UTR und Poly(A)-Schwänzen.
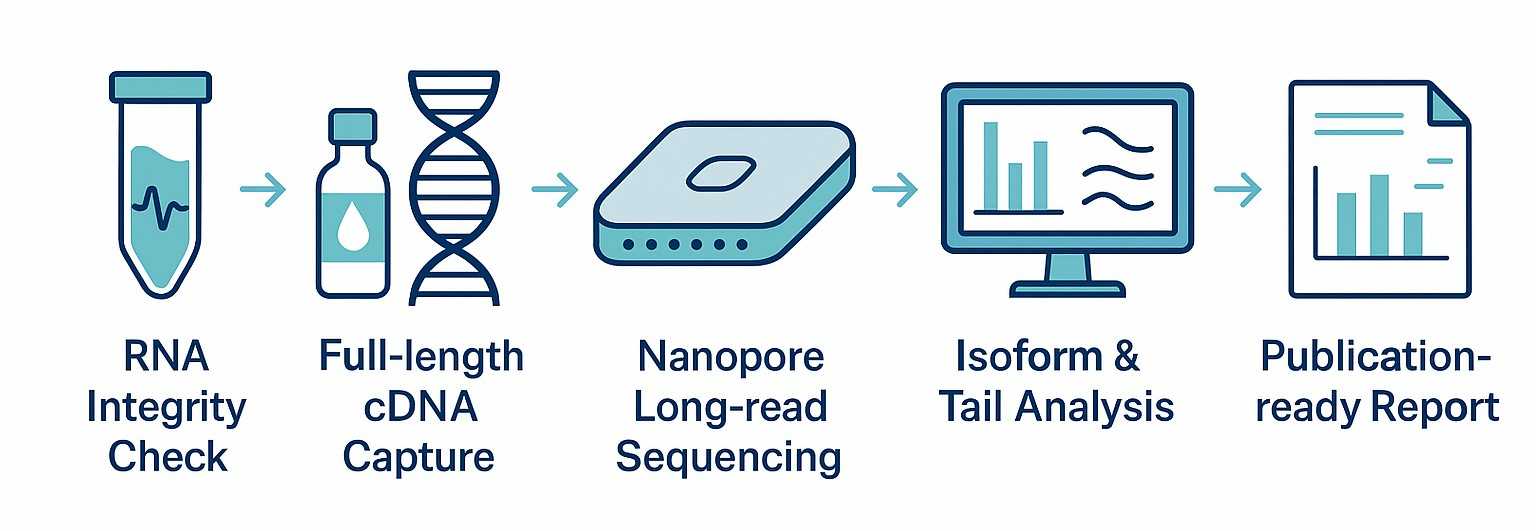 TAIL Iso-seq-Workflow: von RNA-QC, Bibliotheksvorbereitung und Nanopore-Langsequenzierung bis hin zu umfassender bioinformatischer Analyse und publikationsfertigen Berichten.
TAIL Iso-seq-Workflow: von RNA-QC, Bibliotheksvorbereitung und Nanopore-Langsequenzierung bis hin zu umfassender bioinformatischer Analyse und publikationsfertigen Berichten.
Poly(A)-Schwanz-Sequenzierungsmethoden – Wann welche verwenden
| Methode | Plattform | Was es misst | Nicht-A-Reste | Durchsatz / Kosten (relativ) | Typische Stärken | Typische Einschränkungen | Gut für |
|---|---|---|---|---|---|---|---|
| TAIL-seq | Illumina (3'-End-Sequenzierung) | Genomweite Schwanzlänge an den 3'-Enden; 3'-Terminome | Erkennt terminale U/G-Zusätze | Hoch / $$ | Empfindliche Kartierung der 3'-Enden; verknüpft die Schwanzlänge mit dem Zerfall | Rohsignalverarbeitung und benutzerdefinierte Analyse erforderlich; keine vollständigen Isoformen | Deadenylierungs-/Uridylierungsstudien; Stabilitätstests |
| PAL-seq | Illumina + Fluoreszenzstandards | Schwanzlänge mit kalibrierter Fluoreszenz; Merkmale des 3'-Endes | Indirekt | Hoch / $$ | Interne Standards für kalibrierte Schwanzlängen; robuste Bulk-Profilierung | Hinzugefügte Biochemie; spezialisierte Einrichtung; nicht vollständige Länge | Kalibrierte Verteilungen der Schwanzlängen von Bulk; Methodenbenchmarking |
| Poly(A)-Seq | Illumina (direkte Ablesung von poly(A) in cDNA) | Globale Profilierung von Poly(A)-Schwänzen; medianer Schwanz pro Gen | Nicht primärer Fokus | Hoch / $ | Einfachere Bibliothek vs. TAIL-seq; skalierbare Kohorten | Isoformen nicht aufgelöst; Herausforderungen bei Homopolymeren | Großkohorten-Screening von Veränderungen der Schwanzlänge |
| FLAM-seq | PacBio (Langlese cDNA) | Vollständige mRNA einschließlich Poly(A)-Schwanz | Berichtet über nicht-A-Reste | Mäßig / $$–$$$ | Links Isoformstruktur + Schwanzlänge pro Molekül | PacBio-Zugang; längere Laufzeiten | Isoform-resolvente Schwanzbiologie; Schwanzzusammensetzung pro Isoform |
| PAIso-seq / PAIso-seq2 | PacBio HiFi | Transkriptom-weite Schwanzlänge von geringer Input; Vollformat | Kann die Schwanzzusammensetzung erfassen. | Mäßig / $$–$$$ | Hochgenaue HiFi-Lesungen; niedrige Eingabe (z. B. Oocyten) | Plattform-spezifisch; Methodenkomplexität | Wertvolle/niedrig-input Proben; genaue Schwanzschätzungen |
| FLEP-seq2 | Nanopore (Langlese cDNA) | Vollständige RNA mit Schwanzlängen über Gewebe/Spezies hinweg | Erkennt Schwanzmuster | Moderat / $$ | Tiefe-freundlich; kreuzgewebliche Atlanten; nukleäre vs. zytoplasmatische Vergleiche | Handelskompromisse bei der Basisgenauigkeit von Nanoporen | Gewebeatlanten; Pflanzen und nicht-modellierte Organismen |
| Nano3P-seq | Nanopore (3'-End-Capture-cDNA) | RNA-Häufigkeit + Schwanzdynamik (poly(A) und non-poly(A)) | Erkennt Nicht-A-Rückstände | Moderat / $$ | Erfasst polyadenylierte und nicht-polyadenylierte RNAs; pro-Molekül Schwänze | End-Erfassungsbias zum 3'-Ende; erfordert Toolchain | Entwicklungskurs; transkriptomweite Schwanzdynamik |
Bioinformatikanalyse
Unsere interne Pipeline integriert mehrere Informationsschichten:
- Isoform-Identifizierung: Entdeckung von Voll-Längen-Isoformen ohne Assemblierungsverzerrung.
- Poly(A)-Schwanzanalyse: Verteilung, mediane Länge und Nachweis von Nicht-A-Resten (U, G, C).
- APA-Kartierung: Identifizierung von Polyadenylierungsstellen (PACs), Varianten der 3'UTR-Länge und APA-Wechselereignissen.
- Korrelationsanalyse: Verknüpfung der Poly(A)-Länge mit der Transkriptabundanz, Stabilität und translationalen Effizienz.
- Funktionale Interpretation: GO- und KEGG-Anreicherung für von APA betroffene Gene, Vorhersagen zum Gewinn/Verlust von miRNA-Bindungsstellen.
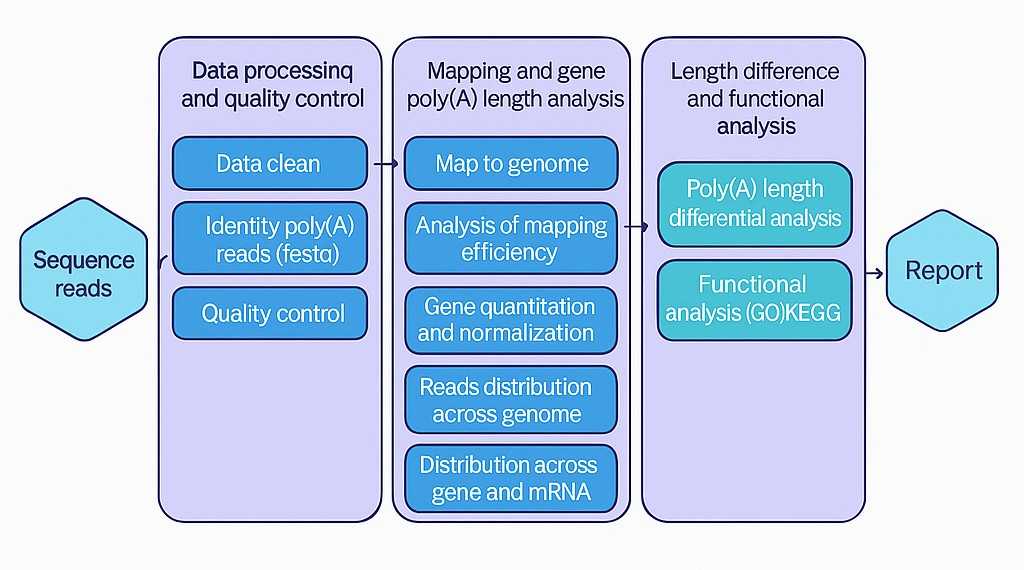
Anwendungen von TAIL Iso-seq
- Dekodierung der Stressreaktion und der Entwicklungsbiologie bei Pflanzen – Profilierung, wie die Poly(A)-Schwanzlänge und die alternative Polyadenylierung (APA) sich unter Hitze, Dürre oder Entwicklungsübergängen (z. B. Arabidopsis-Hitzeschock-PAL-seq-Arbeiten) verändern.
- Verstehen Sie die Krankheitsmechanismen und entdecken Sie Biomarker bei Krebs – identifizieren Sie neuartige Isoformen, veränderte Poly(A)-Schwanzdynamiken oder Fusionstranskripte, die die Pathologie antreiben oder als diagnostische/therapeutische Ziele dienen können (z. B. "Vollständige Länge und Einzelzell-Iso-Seq für die Krebsforschung").
- Optimierung von mRNA-basierten Therapeutika und Impfstoffkonstrukten – Gewährleistung der Transkriptintegrität, Einheitlichkeit der Schwanzlängen und Schwanzzusammensetzung (einschließlich Nicht-A-Resten), um die Translation und Stabilität zu verbessern.
- Entdeckung von Pflanzenmerkmalen und Verbesserung der Züchtung – Nutzung von Schwanzlängen-Signaturen und APA-Mustern zur Verknüpfung mit Ertrag, Hybridvigor, Stressresistenz oder gewebespezifischer Expression in Pflanzen wie Reis, Mais und Sojabohne.
Beispielanforderungen für den TAIL Iso-seq Service
| Probenart | Empfohlene Menge | Mindestmenge | Konzentration / Qualität | Notizen |
|---|---|---|---|---|
| Gesamt-RNA | ≥ 2 µg | 600 ng | ≥ 30 ng/µL | RIN ≥ 8, OD260/280 ≥ 1,8, DNA-frei |
| Zellen | ≥ 1 × 10⁶ | ≥ 5 × 10⁵ | Hohe Lebensfähigkeit (>80%) | Schnellgefrieren von Pellets in flüssigem Stickstoff |
| Gewebe (Tier/Pflanze) | ≥ 50 mg | ≥ 10 mg | RNA-Integrität RIN ≥ 8 | Verunreinigungen entfernen, sofort einfrieren |
| Pflanzengewebe (speziell) | 50–100 mg pro Aliquot | – | RNA-Integrität RIN ≥ 8 | Mit DEPC-Wasser waschen, in flüssigem Stickstoff einfrieren. |
| Bakterienkultur | ≥ 1 × 10⁷ Zellen | – | RNA-Integrität RIN ≥ 8 | Pellet, mit PBS waschen, in flüssigem Stickstoff einfrieren |
Allgemeine Richtlinien
- Reichen Sie Proben in RNase-freiem Wasser oder 10 mM Tris (pH 8,0) ein.
- Vermeiden Sie wiederholte Frost-Tau-Zyklen.
- Versenden Sie Proben auf Trockeneis, um die RNA-Qualität zu erhalten.
- Kennzeichnen Sie die Röhrchen deutlich mit kurzen Codes (≤4 alphanumerische Zeichen).
- Bitte reichen Sie die QC-Daten (Bioanalyzer/Tapestation-Traces) zusammen mit dem Einreichungsformular ein.
Liefergegenstände
- Roh- und verarbeitete Sequenzierungsdaten (FASTQ, BAM).
- Isoform-Level-Transkriptom-Katalog.
- Poly(A)-Schwanzlängenverteilungen und APA-Stellenkarten.
- Weganreicherungs-Tabellen und integrative Visualisierungen.
- Veröffentlichungsbereite Abbildungen und ein detaillierter technischer Bericht.
TAIL Iso-seq häufig gestellte Fragen
Was ist TAIL Iso-seq und wie unterscheidet es sich von regulärem Iso-seq oder RNA-seq?
TAIL Iso-seq ist ein Long-Read-Sequencing-Service (auf Nanopore-Basis), der vollständige Transkripte einschließlich des nativen Poly(A)-Schwanzes erfasst und eine direkte Messung der Schwanzlänge, APA (alternative Polyadenylierung) -Stellen und Isoformvielfalt ermöglicht. Reguläres RNA-seq verliert oft Schwanzinformationen und erfordert eine Lesekonstruktion, während standardmäßiges Iso-seq möglicherweise die Transkriptstruktur erfasst, jedoch nicht immer die vollständige Poly(A)-Zusammensetzung und nicht-A-Reste.
Kann TAIL Iso-seq Nicht-A-Nukleotide innerhalb oder am Ende von Poly(A)-Schwänzen nachweisen?
Ja. Der Service umfasst die Erkennung von Nicht-Adenosin-Resten (wie U, G, C) sowohl an der terminalen Position als auch intern im poly(A)-Schwanz. Diese Nicht-A-Reste geben Aufschluss über die Stabilität des Transkripts, den Abbau oder die Regulierung der Deadenylierung. Werkzeuge wie tailfindr, nanopolish und andere unterstützen diese Analyse.
Wie genau ist die Messung der Poly(A)-Schwanzlänge mit Nanopore-Langlesungen?
TAIL Iso-seq verwendet modernste Basenaufruf- und Signalverarbeitungs-Pipelines, um die Schwanzlänge pro Molekül mit hoher Auflösung zu schätzen, oft mit einer Genauigkeit auf Einzel-Nukleotid-Ebene für kürzere Schwänze und guter relativer Genauigkeit für längere Schwänze. Die Genauigkeit hängt auch von der Probenqualität, dem Sequenzierungsausstoß (.fast5-Retention) und der Werkzeugwahl (tailfindr, nanopolish usw.) ab.
Benötige ich ein Referenzgenom, um TAIL Iso-seq zu verwenden?
Nein, aber das Vorhandensein eines Referenzgenoms oder einer Transkriptannotierung verbessert die Kartierung, die Identifizierung von APA-Stellen und die Isoformauflösung. Bei Nicht-Modellorganismen ist eine de novo-Langleseanordnung in Kombination mit referenzfreier Transkriptannotierung möglich, obwohl einige Analysen (z. B. die Vorhersage von miRNA-Bindungen) ohne gute Annotation schwieriger sein können.
Was sind die Anforderungen an die Eingabemuster für TAIL Iso-seq?
Es wird RNA von hoher Integrität benötigt (empfohlener RIN ≥ 7), frei von Verunreinigungen, mit ausreichender Menge, abhängig von Organismus / Gewebe; geringere Eingaben sind mit optimierten Methoden möglich. Die RNA muss intakte poly(A)-Schwänze und minimale Degradation für die besten Ergebnisse enthalten.
Wie gehen Sie mit PCR-Bias oder der Verkürzung von Poly(A)-Schwänzen in der Pipeline um?
Wir minimieren Verzerrungen, indem wir Long-Read-Sequenzierung verwenden (die die Fragmentzusammenstellung vermeidet), vollständige Transkripte erfassen, Rohsignaldaten (.fast5) zur Schätzung der Schwanzlängen beibehalten und QC-Schritte anwenden, um verkürzte oder degradierte RNA herauszufiltern. Der Einsatz von Werkzeugen, die mit Rohsignalen arbeiten, hilft, eine genaue Schwanzschätzung sicherzustellen.
Was ist der Unterschied zwischen der Analyse des Poly(A)-Schwanzes und der Analyse der alternativen Polyadenylierung (APA)?
Die Analyse des Poly(A)-Schwanzes konzentriert sich auf die Länge, Zusammensetzung und Modifikationen des Schwanzes selbst am Ende des Transkripts. Die APA-Analyse befasst sich damit, wo die Polyadenylierung erfolgt – d.h. das Kartieren von Poly(A)-Schnittstellen, die unterschiedlichen Längen der 3' untranslatierten Regionen (3'UTR) zwischen Isoformen und wie dies die Regulation beeinflusst (z.B. miRNA-Bindung). Beide sind komplementär.
Kann TAIL Iso-seq für vergleichende Studien über Bedingungen oder Arten hinweg verwendet werden?
Ja. Da es die Verteilungen der Schwanzlängen pro Isoform und die Nutzung von APA-Stellen pro Probe bereitstellt, unterstützt TAIL Iso-seq Vergleiche zwischen Geweben, Behandlungen oder Arten. Statistische Pipelines können auf Unterschiede in der Schwanzlänge oder der APA-Nutzung zwischen Gruppen testen.
Wie lange werde ich die Ergebnisse (Bioinformatik + Liefergegenstände) erhalten?
Typische Zeitrahmen hängen von der Anzahl der Proben, der Komplexität und der Sequierungstiefe ab. Der Bericht umfasst rohe und verarbeitete Sequierungsdaten, Verteilungen der Schwanzlängen, APA-Kartierungen, Isoformkataloge und Visualisierungen. (Spezifische Bearbeitungszeiten werden basierend auf dem Umfang im Projektangebot bereitgestellt.)
Wie skaliert die Kosten mit der Anzahl der Proben, der Tiefe oder der Komplexität des Zieltranskripts?
Die Kosten steigen in der Regel mit der Anzahl der Proben, der erforderlichen Lesetiefe und der Komplexität (z. B. Nicht-Modellorganismen oder Transkripte mit geringer Expression). CD Genomics bietet skalierbare Preise an; die Kunden können je nach benötigter Auflösung (Isoformentdeckung, Schwanzzusammensetzung, APA-Präzision) zwischen Standard- oder Premium-Analyseebenen wählen.
TAIL Iso-seq Fallstudien
Zitat: Jia J., Lu W., Liu B. u. a. Ein Atlas der pflanzlichen Voll-Längen-RNA zeigt gewebespezifische und zwischen Monokotylen und Dikotylen konservierte Regulation der Poly(A)-Schwanzlänge., Natur Pflanzen 2022.
1. Hintergrund
Forscher fehlte eine umfassende, vollständige Transkriptomressource in Pflanzen, die umfasst Information zur Länge des poly(A)-Schwanzes über Gewebe und Arten hinweg. Frühere Methoden mit kurzen Reads konnten oft die poly(A)-Schwänze nicht erhalten oder verbanden die Schwänze nicht mit vollständigen Transkript-Isoformen. Diese Lücke schränkte die Erkenntnisse darüber ein, wie die Länge der poly(A)-Schwänze, APA (alternative Polyadenylierung) und die mRNA-Stabilität zwischen Geweben oder über Pflanzenarten hinweg variieren.
2. Methoden
- Verwendete eine poly(A)-anreicherungsfreie, Nanopore-basierte Methode (FLEP-seq2), um vollständige RNAs mit poly(A)-Schwänzen zu sequenzieren.
- Sieben verschiedene Gewebe von Arabidopsis thaliana sowie Sprossgewebe von Mais, Sojabohne und Reis wurden entnommen. Insgesamt wurden über 120 Millionen polyadenylierte mRNA-Moleküle sequenziert.
- Verglich die Verteilungen der Poly(A)-Schwanzlängen zwischen Nukleus und Zytoplasma, verschiedenen Geweben und orthologen Genen in verschiedenen Arten. Auch die Beziehung zwischen der mRNA-Halbwertszeit und der Schwanzlänge wurde bewertet.
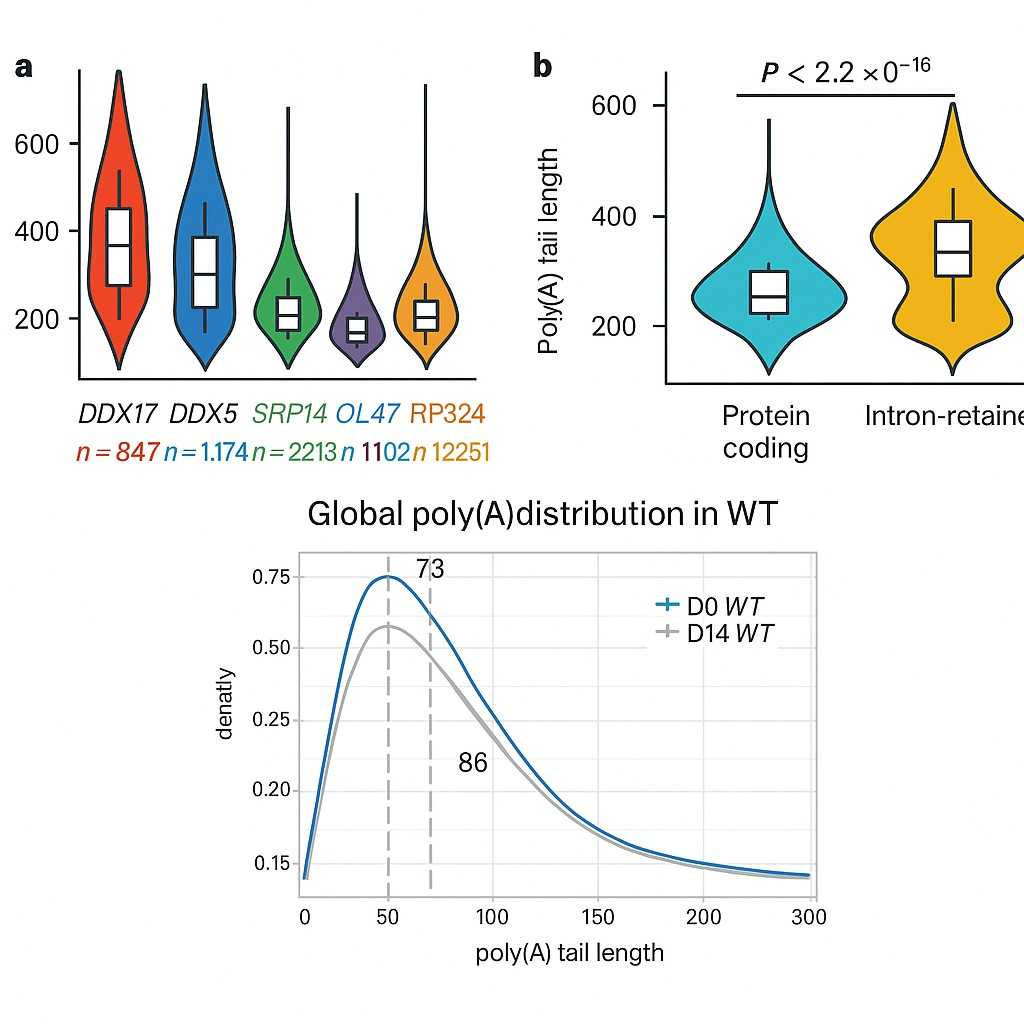
3. Ergebnisse
- In den meisten Geweben erreichten die Längen der Poly(A)-Schwänze Spitzen von etwa ~20 nt und ~45 nt; im Pollen waren die Spitzen unterschiedlich (~55–80 nt).
- Nukleare RNAs hatten Schwänze, die fast doppelt so lang waren wie die Schwänze der zytoplasmatischen RNAs.
- Gene mit kurzen mRNA-Halblebensdauern hatten im Allgemeinen längere Poly(A)-Schwänze, während stabile Transkripte Schwänze hatten, die bei etwa 45 nt ihren Höhepunkt erreichten.
- Der Vergleich zwischen Arten zeigte, dass orthologe Gene dazu neigen, eine ähnliche Regulierung der Schwanzlängen beizubehalten, trotz der Unterschiede zwischen den Arten.
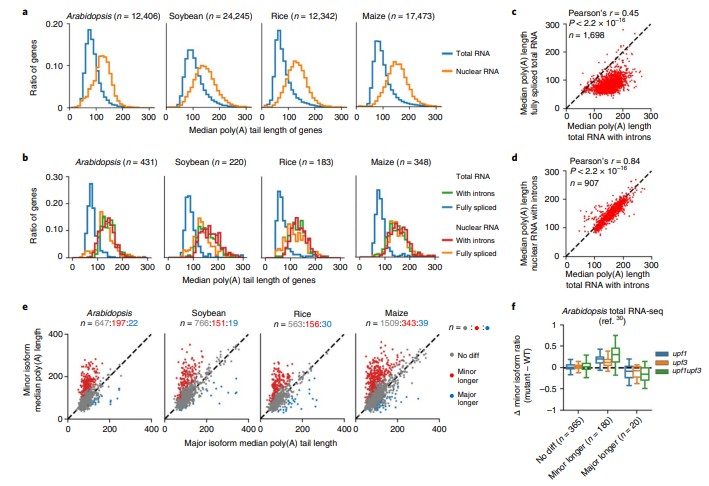 Abbildung: Nukleäre Poly(A)-Schwänze sind länger als zytoplasmatische SchwänzeDies zeigt die Verteilung der Poly(A)-Schwanzlängen in nuklearen vs. zytoplasmatischen Fraktionen über Pflanzenarten/-gewebe hinweg.
Abbildung: Nukleäre Poly(A)-Schwänze sind länger als zytoplasmatische SchwänzeDies zeigt die Verteilung der Poly(A)-Schwanzlängen in nuklearen vs. zytoplasmatischen Fraktionen über Pflanzenarten/-gewebe hinweg.
4. Schlussfolgerungen
- Die Regulation des Poly(A)-Schwanzes ist genspezifisch, gewebespezifisch und zeigt eine evolutionäre Erhaltung unter Pflanzen.
- Unterschiede in der Schwanzlänge zwischen Zellkern und Zytoplasma sowie zwischen Geweben deuten auf eine dynamische Regulation des Schwanzabschneidens (Deadenylierung) und der APA hin.
- Der Datensatz bietet eine Ressource zum Studium der mRNA-Stabilität, Translation, APA und nicht-kodierender regulatorischer Merkmale in Pflanzen.
- Unterstützt den Wert der vollständigen, langformatigen Sequenzierung, um die Schwanzlänge mit der RNA-Funktion zu verknüpfen.
Referenzen:
- Jia J, et al. Ein Atlas der vollständigen RNA von Pflanzen zeigt die gewebespezifische und die bei Monokotylen und Dikotylen konservierte Regulation der Poly(A)-Schwanzlänge.. Natur Pflanzen. 2022
- Wen H, Chen W, Chen Y, Wei G, Ni T. Integrative Analyse von Iso-Seq und RNA-seq zeigt dynamische Veränderungen von alternativen Promotoren, alternativer Spleißung und alternativer Polyadenylierung während der durch Angiotensin II induzierten Seneszenz in primären Ratten-Aortenendothelzellen.. Front Genet. 2023 Jan 19;14:1064624.
- Kuo RI, Cheng Y, Zhang R, Brown JWS, Smith J, Archibald AL, Burt DW. Die dunkle Seite des menschlichen Transkriptoms mit Langlese-Transkript-Sequenzierung erhellen. BMC Genomik2020, 30. Oktober; 21(1):751. doi: 10.1186/s12864-020-07123-7. PMID: 33126848; PMCID: PMC7596999.


