
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Mikrosatelliten-Genotypisierungsdienst
CD Genomics verfügt über umfangreiche Erfahrung in der Unterstützung bei der Auswahl und dem Design von Mikrosatellitenmarkern für eine Vielzahl von Pflanzen- und Tierarten. Neben der Mikrosatellitengenotypisierung bieten wir auch an Genotypisierung durch Sequenzierung (GBS) Methode, die Zeit und Ressourcen spart.
Die Einführung der Mikrosatelliten-Genotypisierung
Mikrosatelliten, auch bekannt als einfache Sequenzwiederholungen (SSRs) oder kurze tandemartige Wiederholungen (STRs), sind aufgrund ihres hohen Polymorphismus beliebte Marker. Genotypisierung ist ein genauer, kosteneffektiver und schneller Ansatz zur Unterscheidung von Mikrosatellitenallelen, der durch fortlaufende technische Fortschritte, einschließlich Multiplex-PCR, unterstützt wird und Next-Generation-Sequenzierung Technologien. Der vollständige Workflow für die Mikrosatelliten-Genotypisierung umfasst die Erfassung von Sequenzierungsdaten, die Auswahl von SSRs, das Design von Primern, die Validierung von Primern, die Multiplex-PCR und die kapillare Gelelektrophorese in Verbindung mit einer fluoreszenzbasierten Detektion sowie die Datenanalyse.
Die PCR-Reaktion wird mit fluoreszenzmarkierten Primern durchgeführt, anschließend können die PCR-Fragmente in einer Kapillare analysiert werden. DNA-Sequenzierung Maschine, und die Daten werden mit GeneMapper analysiert.TM Software. Durch die Nutzung von Multiplexing nach Fragmentgröße und Farbstofffarbe sowie der Fähigkeit automatisierter fluoreszierender genetischer Analysegeräte, 16 Proben gleichzeitig aus 96-Well- oder sogar 384-Well-Mikrotiterplatten zu laden, kann eine Hochdurchsatz-Markergenanalyse entworfen werden. Dieser Service wurde auf eine Vielzahl von Arten angewendet und kann für jede Art genutzt werden, für die Mikrosatellitenmarker verfügbar sind.
Wenn Sie mehr über Mikrosatelliten-Genotypisierung erfahren möchten, können Sie unseren Artikel lesen "Einführung in Mikrosatelliten und Mikrosatelliten-Genotypisierung".
Vorteile der Mikrosatelliten-Genotypisierung
Vorteile von Mikrosatelliten als genetische Marker:
- Universell und polymorph.
- Locus-spezifisch (im Gegensatz zu Multi-Locus-Markern wie Minisatelliten)
- Kodominant (Heterozygoten können von Homozygoten unterschieden werden)
- PCR-basiert (benötigt nur winzige Mengen Gewebe und funktioniert mit degradiertem DNA)
- Mehrere Anwendungen (von individueller Identifikation bis hin zu feinkalibrigen Phylogenien)
Anwendung der Mikrosatelliten-Genotypisierung
Die riesige Menge an Daten, die für Tausende von Mikrosatellitenmarkern über Organismen hinweg entsteht, macht die Mikrosatelliten-Genotypisierung zu einem weithin akzeptierten Werkzeug für:
- Verknüpfungsanalyse der Ko-Segration
- Diagnose und Identifizierung menschlicher Krankheiten
- Forensische Identifikation und Verwandtschaftstests
- Zelllinienidentifikation
- Bevölkerungsstudien
Mikrosatelliten-Genotypisierungs-Workflow
CD Genomics nutzt fortschrittliche Plattformen (ABI 3730xl DNA-Analyzer) und Strategien (Multiplex-PCR), um einen schnellen und genauen Mikrosatelliten-Genotypisierungsservice sowie bioinformatische Analysen anzubieten. Unser hochqualifiziertes Expertenteam führt ein Qualitätsmanagement durch und befolgt jeden Schritt, um zuverlässige und unvoreingenommene Ergebnisse zu gewährleisten. Der grobe Arbeitsablauf ist unten skizziert.
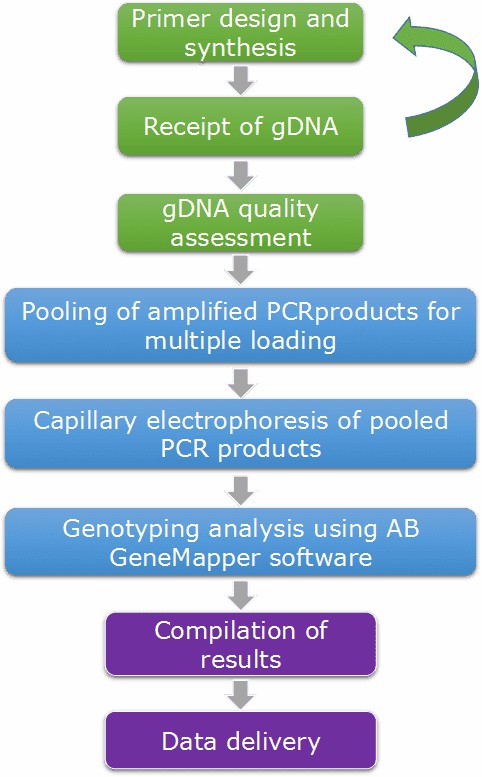
Dienstspezifikation

|
Musteranforderungen
|
 |
Multiplex-PCR und Genotypisierung
|

|
Datenanalyse
|
Analyse-Pipeline
Liefergegenstände
- Vollständiges experimentelles Verfahren,
- PCR-Elektrophorese-Bilder
- Große Gel-Detektionsbilder
- Sequenzierungschromatogramme
- Ergebnisse der Datenanalyse
CD Genomics bietet ein umfassendes Mikrosatelliten-Genotypisierungsdienstleistungspaket an, das das Entwerfen und Bestellen von fluoreszenzmarkierten Primerpaaren, die Primervalidierung, die Mikrosatelliten-Genotypisierung sowie die Datenanalyse umfasst. Wir können diesen Ablauf an Ihr Forschungsinteresse anpassen. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Demonstrationsergebnisse
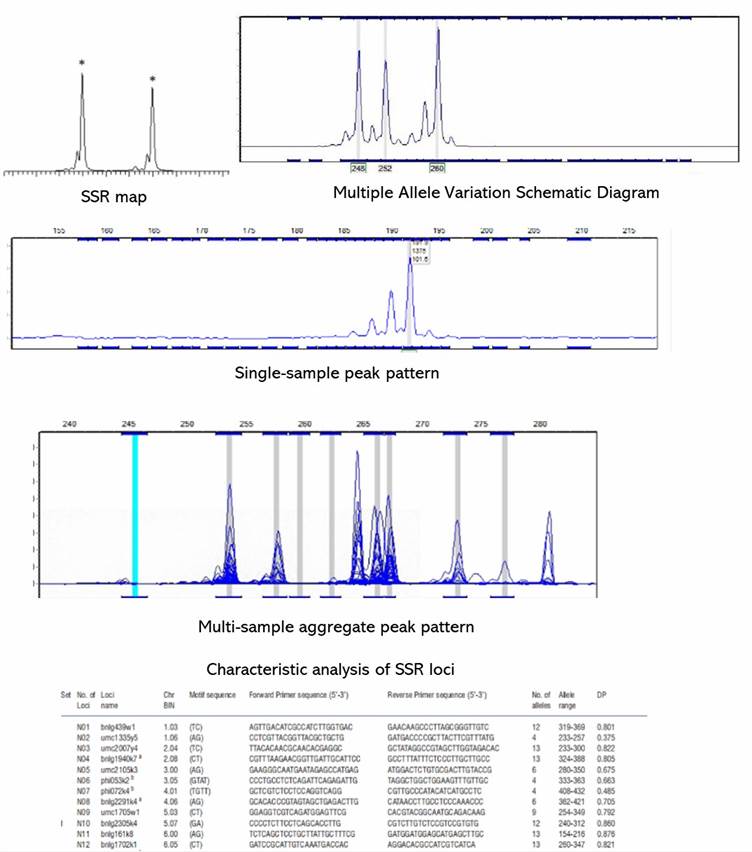
FAQs zum Mikrosatelliten-Genotyping-Service
Was ist Mikrosatelliten-Genotypisierung?
Mikrosatelliten-Genotypisierung ist eine leistungsstarke Technik, die zur Erkennung und Bewertung von Variationen innerhalb von Mikrosatelliten, auch bekannt als einfache Sequenzwiederholungen (SSRs), in DNA eingesetzt wird. Mikrosatelliten sind kurze sich wiederholende Sequenzen, die aus 1-6 Basenpaaren bestehen. Diese Sequenzen sind im gesamten Genom erheblich variabel und dienen daher als ausgezeichnete genetische Marker.
2. Was ist das Prinzip der Mikrosatelliten-Genotypisierung?
Die Mikrosatelliten-Genotypisierung basiert auf dem Prinzip der Polymerase-Kettenreaktion (PCR) zur Amplifikation spezifischer Mikrosatellitenstandorte. Diese Technik umfasst das Design spezifischer Primer zur Amplifikation gezielter Mikrosatellitenregionen. Anschließend werden die amplifizierten Produkte anhand von Größenvariationen durch Techniken wie Gelelektrophorese oder Kapillarelektrophorese differenziert und typisiert.
3. Was sind die Vorteile der Mikrosatelliten-Genotypisierungstechnologie?
Die Verwendung von Mikrosatelliten-Genotypisierungstechnologie bietet mehrere deutliche Vorteile in Studien zur genetischen Variation:
- Hervorstechender Polymorphismus: Mikrosatelliten zeichnen sich durch ihren hohen Variabilitätsgrad aus und liefern somit eine Fülle genetischer Informationen.
- Präzise Auflösung: Diese Untersuchungstechnologie ermöglicht eine hochpräzise Differenzierung und hebt selbst kleinste genetische Variationen zwischen einzelnen Organismen hervor.
- Breite Verteilung: Mikrosatellitenmarker durchdringen das Genom umfassend, was diese Technik für verschiedene biologische Arten geeignet macht.
4. Wie unterscheidet sich die Mikrosatelliten-Genotypisierung von anderen Genotypisierungstechnologien?
Mikrosatelliten-Genotypisierung unterscheidet sich von Einzelne Nukleotid-Polymorphismus (SNP) Genotypisierung in mehreren entscheidenden Aspekten:
- Erweiterter Polymorphismus: Im Vergleich zu SNP-GenotypisierungDie Mikrosatelliten-Genotypisierung weist ein überlegenes Maß an Polymorphismus auf, was sie außergewöhnlich gut für die Erforschung hochvariabler genetischer Regionen geeignet macht.
- Unterschiedliche Anwendungszwecke: Während SNP-Genotypisierung traditionell bei der Analyse von Punktmutationen nützlich ist, glänzt die Mikrosatelliten-Genotypisierung in Studien, die ein detaillierteres Verständnis der genetischen Variabilität erfordern.
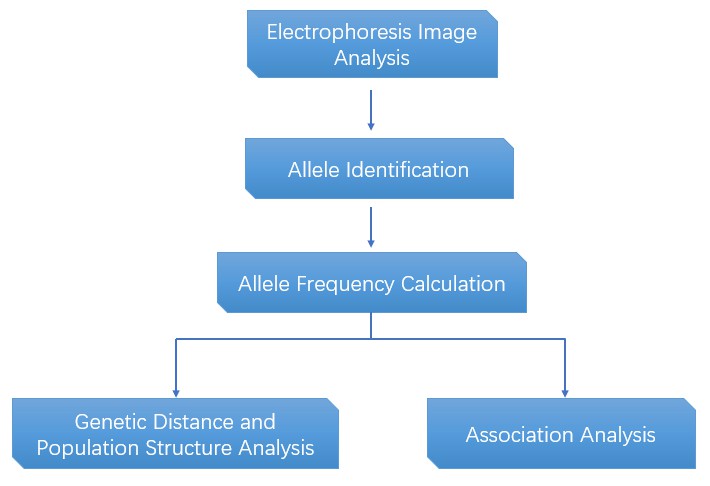
5. Wie können Mikrosatelliten-Genotypisierungsdaten verarbeitet und analysiert werden?
Die Verfahren zur Verarbeitung und Analyse von Mikrosatelliten-Genotypisierungsdaten umfassen:
- Fragmentgrößenermittlung: Die Messung der Fragmentgrößen aus Elektropherogrammen erfolgt mit spezieller Software.
- GenotypisierungBasierend auf der Fragmentgröße erfolgt die Identifizierung von Allelen an Mikrosatellitenloci.
- Statistische Analyse: Genotypische Daten werden mit statistischer Software analysiert, um genetische Variabilität und Populationsstruktur aufzudecken.
Mikrosatelliten-Genotypisierungsdienst Fallstudien
Hochauflösende, hochgenaue Mikrosatelliten-Genotypisierung ermöglicht eine präzise Risikoklassifikation für Lungenkrebs.
Journal: Onkogen
Impact-Faktor: 7,519
Veröffentlicht: 31. Juli 2017
Hintergrund
Mikrosatelliten sind eine Reihe von tandemartig angeordneten DNA-Sequenzen, die aus kurzen repetitiven Einheiten (1-6 bp) bestehen. Jüngste Forschungen haben die entscheidende Rolle hervorgehoben, die diese Mikrosatelliten im komplexen Prozess der genetischen Entwicklung bei einer Vielzahl von Krebsarten spielen. Das Hauptziel dieser Studie besteht darin, bestimmte Mikrosatellitenmarker zu identifizieren, die für die Risikostratifizierung bei Lungenkrebs anwendbar sind. Dies soll erreicht werden, indem die Keimbahn-Exomsequenzen von Lungenkrebspatienten mit einer normativen Kontrollgruppe verglichen werden.
Methoden
- 266 LUAD- und 222 LUSC-Germline-Exomproben
- 1kGP Nicht-Tumor-Kontrollproben
- Genomische DNA-Extraktion
- Zielanreicherung
- Zielsequenzierung
- Genotypisierung
- ROC-Kurvenanalyse
- Mechanistische Analyse
- Weganalyse
Ergebnisse
Die angewandte Methodik ist hauptsächlich in zwei Phasen unterteilt: (i) Identifizierung von Mikrosatelliten (MST) Standorten und (ii) Validierung ihres Status als signifikante Marker. Unter Verwendung von Daten aus dem Cancer Genome Atlas (TCGA), bestehend aus 488 Tumormustern von nicht-kleinzelligem Lungenkrebs, und aus dem 1000 Genomes Project mit 399 Kontrollproben ohne Krebs, konzentrierte sich die erste Phase auf die Unterscheidung genetischer Variationen an MST-Standorten. Diese Analyse führte zur Enthüllung von 119 signifikanten MST-Standorten, was auf einen entscheidenden Genotypunterschied zwischen krebsartigen und Kontrollproben hindeutet. Durch ähnliche Analysemethoden wurden zusätzlich 144 MST-Marker in anderen Krebsarten identifiziert. Der Übergang zur zweiten Phase beinhaltete die Entwicklung eines maßgeschneiderten Zielanreicherungs-Kits mit 263 (119+144) MST-Markern, ergänzt durch 84 Kontroll-MST-Marker für die gezielte Erfassung von Sequenzen. Die Durchführung einer Tiefensequenzierung von 30 Lungenkrebsproben und 89 Kontrollproben ohne Krebs, mit einer durchschnittlichen Sequenzierungstiefe von 579x ± 315, offenbarte ausgeprägte Unterschiede innerhalb von 21 (13+8) MST-Markern unter den 263, die untersucht wurden.
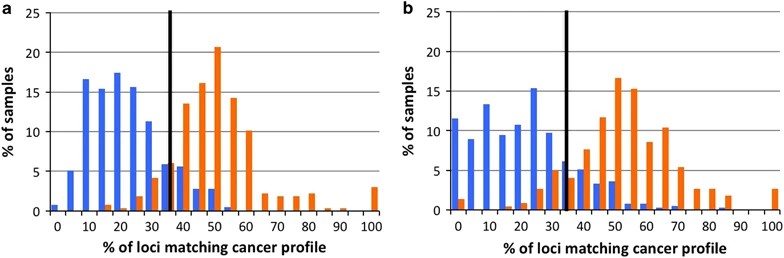 Abb. 1. Die rechnerisch ermittelten MST-Loci von LUAD und LUSC unterscheiden ihren jeweiligen Krebs-Typ mit hoher Sensitivität von 1000 Genomen Nicht-Krebs-Kontrollproben (LUAD: 0,87, LUSC: 0,88).
Abb. 1. Die rechnerisch ermittelten MST-Loci von LUAD und LUSC unterscheiden ihren jeweiligen Krebs-Typ mit hoher Sensitivität von 1000 Genomen Nicht-Krebs-Kontrollproben (LUAD: 0,87, LUSC: 0,88).
Jüngste Studien heben hervor, dass verschiedene Krebsarten tatsächlich gemeinsame onkogene Merkmalspektren teilen können. Neben der Entdeckung von 13 Mikrosatelliten (MST)-Standorten, die zur Risikostratifizierung von Lungenkrebs beitragen, wurden in dieser Studie auch 144 MST-Standorte aus anderen Krebsarten, einschließlich Brustkrebs, Eierstockkrebs, Melanomen und Neuroblastomen, untersucht. Nach rigoroser Überprüfung wurde festgestellt, dass 8 dieser Marker effektiv im Rahmen der Risikobewertung für Lungenkrebs genutzt werden können.
Tabelle 1 MST-Loci, die zwischen den Lungenkrebsproben und den Nicht-Tumorproben präzise unterscheiden können.
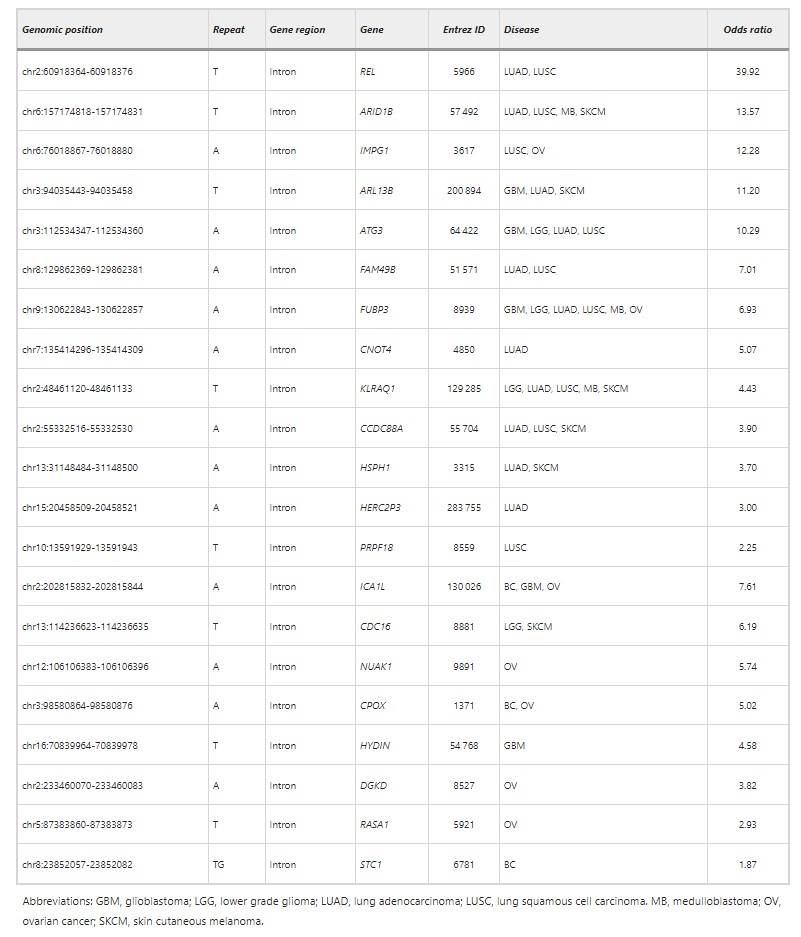
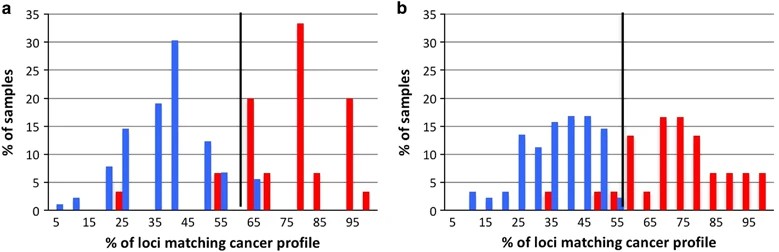 Abb. 2. Die 13 spezifischen MST-Loci für Lungenkrebs und 8 MST-Loci, die spezifisch für andere Krankheiten sind, können zwischen den Lungenkrebs- und den nicht-krebsartigen Kontrollgruppen unterscheiden.
Abb. 2. Die 13 spezifischen MST-Loci für Lungenkrebs und 8 MST-Loci, die spezifisch für andere Krankheiten sind, können zwischen den Lungenkrebs- und den nicht-krebsartigen Kontrollgruppen unterscheiden.
Eine genauere Untersuchung dieser 13 MST-Stellen ergab deren Lage im Intronbereich von Genen. Frühere Untersuchungen hatten die Hypothese aufgestellt, dass MST im Intronbereich den Prozess der Splice-Variante und die Transkription von Genen beeinflussen könnte. Eine Clusteranalyse dieser 13 Gene innerhalb der DAVID-Datenbank zeigte eine Momentaufnahme einer signifikanten Anreicherung sowohl in Splice-Varianten als auch in Transkriptprodukten. Eine Assoziationsanalyse dieser 13 Gene, die mit Lungenkrebs in Verbindung stehen, ergab, dass nur REL und ARID1B wurden in früheren Forschungen als mit Krebs in Verbindung stehend identifiziert. Die Implikationen der TCGA deuten darauf hin, dass diese beiden Gene das Auftreten von Lungenkrebs durch Mechanismen von DNA-Schäden und Chromatinumbau begünstigen könnten.
Fazit
- Eine umfassende Analyse von krebsbezogenen Daten aus dem TCGA und dem 1000 Genomes Project erfasste 263 (119+144) MST-Stellen, die signifikante Genotypvariationen zwischen Krebs- und Kontrollproben aufwiesen.
- Durch den Einsatz von gezieltem Capture-Sequencing wurde eine effektive Probenvalidierung für diese MST-Stellen durchgeführt, die 21 (13+8) MST-Marker enthüllte, die eine verbesserte Risikostratifizierung für Lungenkrebs ermöglichen.
- Die Analyse der 13 gesichteten MST-Standorte durch Cluster- und Anreicherungsanalysen ergab, dass sie möglicherweise DNA-Schäden und Chromatin-Remodeling-Mechanismen beeinflussen, was folglich zu Lungenkrebs führt. Dieser Effekt erfolgt überwiegend durch REL und ARID1B.
- Die unentdeckten MST-Stätten haben potenziell erheblichen klinischen Wert. Ihre Entdeckung ist von entscheidender Bedeutung für die Identifizierung neuer therapeutischer Ziele, die Vorhersage von Lungenkrebs und wesentliche Prozesse zur Bewertung des Krebsrisikos.
Referenz:
- Velmurugan KR, Varghese RT, Fonville NC, u. a.Hochpräzise Mikrosatelliten-Genotypisierung mit hoher Tiefe und Genauigkeit ermöglicht eine präzise Risikoklassifizierung für Lungenkrebs. Onkogen2017, 36(46):6383-90.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Pilze: Freunde oder Feinde – eine wissenschaftliche Outreach-Initiative zur Sammlung von luftgetragenen Pilzsporen durch Schüler der Oberstufe
Zeitschrift: Zeitschrift für Mikrobiologie und Biologiedidaktik
Jahr: 2024
Kleine, aber signifikante genetische Differenzierung zwischen Populationen von Phyllachora maydis im mittleren Westen der Vereinigten Staaten, aufgedeckt durch Mikrosatelliten (SSR) Marker.
Journal: bioRxiv
Jahr: 2023
Das genetische Erbe von Fragmentierung und Übernutzung im bedrohten medizinischen afrikanischen Pfefferbaum, Warburgia salutaris
Journal: Wissenschaftliche Berichte
Jahr: 2020
Bewertung von Plasma-Biomarkern für die A/T/N-Klassifikation der Alzheimer-Krankheit bei Erwachsenen karibisch-hispanischer Ethnie
Journal: JAMA Netzwerk Offen
Jahr: 2023
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


