
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Warum tRNA-Sequenzierung wichtig ist
Während traditionell Transkriptomik und Ribosomen-Profiling haben unser Verständnis der Genexpression transformiert, bleibt die Komplexität der Biologie der Transfer-RNA (tRNA) eine unerforschte Grenze. tRNAs sind essentielle Moleküle, die mRNA-Codons und Aminosäuren verbinden und direkt die Proteinsynthese steuern. Allerdings können subtile Veränderungen in der tRNA-Häufigkeit, chemischen Modifikationen oder dem Ladezustand die translationale Regulation erheblich beeinflussen, was Auswirkungen auf Gesundheit und Krankheit hat.
Modern tRNA-Sequenzierungsmethoden haben neue Möglichkeiten eröffnet, diese Moleküle in bisher unerreichter Detailgenauigkeit zu untersuchen. Durch die Nutzung von Technologien wie tRNA-Sequenzierung Nanopore-Plattformen, Forscher können die analysieren tRNA-Sequenzlängekritische posttranskriptionale Modifikationen erkennen und untersuchen, wie sich tRNA-Populationen als Reaktion auf Stress, Krebsprogression, Stoffwechselstörungen oder Virusinfektionen verändern.
Darüber hinaus Erkenntnisse aus tRNA-Sequenzierungsdienste beitragen zum Aufbau umfassender tRNA-Sequenzdatenbanken, die fortgeschrittene Studien in Genomik, translationaler Medizin und therapeutischer Entwicklung unterstützen. Die Profilierung des tRNAome ist entscheidend geworden, um die zelluläre Anpassung zu entschlüsseln und neuartige Biomarker oder therapeutische Ziele zu entdecken.
In der Forschung und Arzneimittelentwicklung ist die Fähigkeit, die tRNA-Sequenzlänge und die Modifikationslandschaft sorgt für eine präzise Interpretation der zellulären Reaktionen, hilft Missverständnisse zu vermeiden und beschleunigt Entdeckungen.
Serviceübersicht
Bei CD Genomics bieten wir eine umfassende tRNA-Sequenzierungsdienst entworfen, um die Komplexität der translationalen Regulation und der zellulären Anpassung zu beleuchten. Dabei werden sowohl traditionelle tRNA-Sequenzierungsmethoden und innovativ tRNA-Sequenzierung Nanopore-TechnologienWir bieten unvergleichliche Einblicke in die tRNA-Dynamik in verschiedenen biologischen Kontexten.
Unsere Dienstleistungen erfassen wichtige Daten zu:
- tRNA-Häufigkeit: Quantifizieren Sie die Werte über Gewebe, Zelltypen oder experimentelle Bedingungen hinweg.
- tRNA-Modifikationen: Identifizieren und Lokalisieren chemischer Veränderungen, die die Stabilität und Funktion von tRNA beeinflussen.
- tRNA-Sequenzlänge: Vollständige tRNA-Moleküle zur genauen Kartierung und Analyse erkennen.
- Aminoacylierungsstatus: Messen Sie den Beladungszustand von tRNAs, der entscheidend für das Verständnis der Übersetzungseffizienz ist.
Die Expertise von CD Genomics stellt sicher:
- Direkte Sequenzierung von nativer tRNA ohne cDNA oder PCR, Minimierung von Verzerrungen und Erhaltung natürlicher Modifikationen.
- Erstellung zuverlässiger, hochwertiger tRNA-Sequenzdatenbanken für nachgelagerte Forschung.
- Leistungsstarke bioinformatische Lösungen, die publikationsbereite Visualisierungen liefern, einschließlich Vulkanplots, Heatmaps und Streudiagrammen.
Warum CD Genomics für tRNA-Sequenzierung wählen?
✅ Fortgeschrittene tRNA-Sequenzierungsmethoden
Wir bieten sowohl traditionelle Protokolle als auch tRNA-Sequenzierung mit Nanoporen Lösungen, die eine Einzelmolekülauflösung und die direkte Erkennung von nativen Modifikationen ermöglichen, ohne dass eine cDNA-Umwandlung oder PCR erforderlich ist. Dies gewährleistet eine hochgenaue und umfassende tRNA-Profilierung.
✅ Vollständige tRNA-Sequenzierung
Erfassung abgeschlossen tRNA-Sequenzlänge Information für präzise Kartierung und Variantenerkennung, die entscheidend für das Verständnis der translationalen Regulation und die Identifizierung krankheitsspezifischer Veränderungen ist.
✅ Umfassende Änderungsdetektion
Erkennen Sie verschiedene posttranskriptionale Modifikationen, wie Methylierung und Pseudouridylierung, und zeigen Sie, wie diese chemischen Veränderungen die Stabilität, Faltung und Funktion von tRNA beeinflussen.
✅ Aminoacylierungsanalyse
Messen Sie die geladenen vs. ungeladenen tRNA-Pools, um zu untersuchen, wie die Aminoacylierungdynamik die Übersetzungseffizienz beeinflusst – ein Schlüsselfaktor in Krankheitsstudien und therapeutischer Forschung.
✅ Hohe Empfindlichkeit bei niedrigen Eingabebedürfnissen
Unsere optimierten Arbeitsabläufe liefern hochwertige Ergebnisse aus minimalem Ausgangsmaterial, was tRNA-Sequenzierungsdienste zugänglich für wertvolle oder begrenzte Proben.
✅ Umfassende Integration von tRNA-Sequenzdatenbanken
Profitieren Sie von unseren kuratierten und autoritativen tRNA-Sequenzdatenbanken, die die Annotationgenauigkeit verbessern und eine robuste bioinformatische Analyse ermöglichen.
✅ Veröffentlichungsfertige Datenvisualisierungen
Wir bieten hochwertige Abbildungen an, darunter Vulkan-Diagramme, Heatmaps und Streudiagramme, die sich für Veröffentlichungen, Präsentationen oder regulatorische Einreichungen eignen.
✅ Expertenunterstützung in Bioinformatik
Unser Team von Bioinformatik-Spezialisten bietet eine umfassende Datenanalyse und liefert Erkenntnisse, die auf Ihre spezifischen Forschungsziele zugeschnitten sind.
Arbeitsablauf / Wie es funktioniert
Probenvorbereitung
Extraktion und Reinigung von totaler RNA oder kleiner RNA aus verschiedenen Probenarten.
tRNA-Bearbeitung
Deacylierung und Adapterligierung, um eine hohe Effizienz für die Vollständigkeit zu gewährleisten. tRNA-Sequenzlänge fangen.
Bibliotheksbau
Erstellung von Bibliotheken, die für Hochdurchsatz-Sequenzierung oder direkte native RNA-Sequenzierung geeignet sind.
Sequenzierung
Hochauflösende Sequenzierung mit entweder Illumina-Plattformen oder nanopore-basierten Systemen für die direkte RNA-Analyse.
Bioinformatische Analyse
Umfassende Datenverarbeitung, Annotation gegen autoritative tRNA-Sequenzdatenbankenund Erstellung publikationsreifer Visualisierungen.
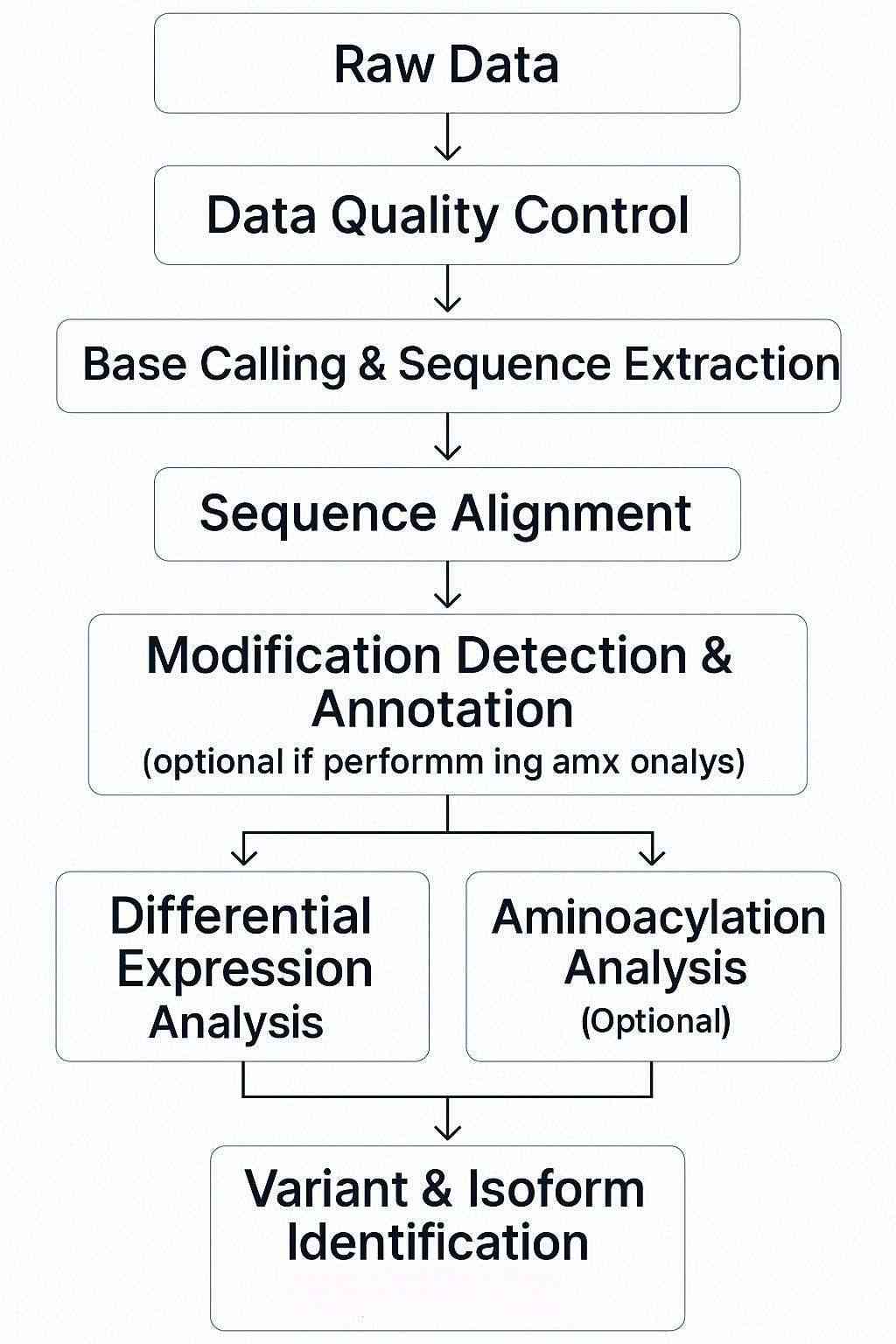
Bioinformatik-Liefergegenstände
Analyse-Pipeline
- Differenzielle Expressionsanalyse
Identifiziert signifikante Unterschiede in der tRNA-Sequenzlänge und -häufigkeit unter verschiedenen Bedingungen. Visuelle Ausgaben umfassen Vulkan-Diagramme, Streudiagramme und Heatmaps. - Änderungserkennung und -zuordnung
Erkennt und annotiert post-transkriptionale Modifikationen innerhalb von tRNA-Sequenzen und zeigt deren funktionalen Einfluss auf die Translation und Krankheitsprozesse. - Aminoacylierungsstatus-Profilierung
Maßnahmen zur Analyse von geladenen vs. ungeladenen tRNA-Pools, die Einblicke in die Übersetzungseffizienz und metabolische Anpassung bieten. - Variant- und Isoform-Identifikation
Erkennt Sequenzvarianten, Mutationen und neuartige Isoformen mithilfe kuratierter tRNA-Sequenzdatenbanken und unterstützt die Forschung zu Krankheitsbiomarkern sowie die therapeutische Entwicklung. - Benutzerdefinierte Berichte
Liefert interaktive, veröffentlichungsbereite Berichte, die für wissenschaftliche Präsentationen, Publikationen oder regulatorische Einreichungen formatiert sind. - Datenintegrationsunterstützung
Integriert tRNA-seq-Ergebnisse mit anderen Omics-Datensätzen (z. B. Transkriptomik, Proteomik) für tiefere biologische Einblicke.
Datenausgabe
- Formate: FASTQ, BAM, VCF, tabulatorgetrennte Ausdruckstabellen
- Visualisierungen: Heatmaps, Streudiagramme, Vulkanplots, Variantenplots
- Annotierungsdateien: GFF3, BED oder benutzerdefinierte Formate auf Anfrage
- Berichte: PDF-Zusammenfassungen, interaktive Excel-Tabellen
Unterstützte Probenarten
- Gesamt-RNA aus Geweben, Zellen, Flüssigkeiten
- Niedrig-input RNA-Proben
- Reinige tRNA-Fraktionen
- Herausfordernde Proben mit hohen Modifikationsgraden
Recheninfrastruktur
- Hochleistungsrechner-Cluster
- Dedizierte Bioinformatik-Pipelines, die optimiert sind für:
tRNA-Sequenzierung Nanopore-Daten
- Traditionelles kurzes Lese-tRNA-seq
- Benutzerdefinierte Analyseumgebungen auf Anfrage verfügbar
Bioinformatik-Expertise
Unterstützung für:
- Beratung zum experimentellen Design
- Statistische Analyse und Interpretation
- Abbildung und Tabellenvorbereitung für die Veröffentlichung
- Spezialisiertes Wissen in der tRNA-Biologie und Modifikationsanalyse
Anwendungen

🔬 Krebsforschung & Tumorbiologie
Untersuchen Sie, wie Veränderungen in tRNA-Sequenzlänge, Fülle und Modifikationen treiben die Onkogenese, Metastasierung und Therapieresistenz voran. Identifizieren Sie potenzielle tRNA-basierte Biomarker für Diagnostik oder therapeutische Ziele.
🧬 Studien zur translationalen Regulation
Verstehen, wie Zellen die Genexpression durch dynamische Veränderungen in tRNA-Populationen, Modifikationen und Aminoacylierungsstatus unter verschiedenen physiologischen oder pathologischen Bedingungen feinabstimmen.
🧪 Biomarker-Entdeckung & Personalisierte Medizin
Hebel tRNA-Sequenzierungsdienst Daten zur Entdeckung neuer Biomarker und zur Entwicklung von Präzisionsmedizin-Strategien, die auf patientenspezifische translational Profile zugeschnitten sind.
🦠 Infektionskrankheiten und Virologie
Untersuchen, wie Viren den Wirt manipulieren tRNA-Sequenzdatenbanken und tRNA-Pools, um die Translation viraler Proteine zu optimieren und immunologische Reaktionen zu umgehen.
🧠 Neurodegenerative und Stoffwechselerkrankungen
Analysiere die Dysregulation von tRNA, die mit Erkrankungen wie der Huntington-Krankheit, Neurodegeneration und Stoffwechsel-Syndromen in Verbindung gebracht wird, um die therapeutische Forschung zu unterstützen.
🧫 Synthetische Biologie & Biotechnologie
Optimierung der Codon-Nutzung und Übersetzungseffizienz durch Mapping tRNA-Sequenzierung mit Nanoporen Daten, Verbesserung von Genbearbeitungsdesigns und synthetischen Konstrukten.
⚙️ Gentherapie und Entwicklung von ATMPs
Gewinnen Sie Einblicke in die tRNA-Dynamik, die entscheidend für die Entwicklung fortschrittlicher therapeutischer Arzneimittel (ATMPs) ist, um die Übersetzungseffizienz und Sicherheitsprofile zu verbessern.
Musteranforderungen
| Probenart | Mindestbetrag | Notizen |
|---|---|---|
| Zellen | 2 × 10⁶ Zellen | Unter RNase-freien Bedingungen geerntet. |
| Gewebe | 50 mg | Frisch oder gefroren; bei -80°C gelagert. |
| Vollblut / Serum / Plasma | 2–3 ml | In EDTA- oder Heparinröhrchen sammeln; bei -80 °C lagern. |
| Hirn-Rückenmarks-Flüssigkeit (HRF) | 5 ml | Bei -80 °C lagern; Gefrier-Tau-Zyklen vermeiden. |
| Urin | 50 ml | Zentrifugieren Sie, um Zellen zu pelleted, falls erforderlich; lagern Sie das Pellet bei -80 °C. |
| Gesamt-RNA | ≥ 2 µg | OD260/280 ≥ 1,8; OD260/230 ≥ 1,5; intakte RNA mit klaren Banden in der Elektrophorese. |
Lager- und Versandempfehlungen:
- Versenden Sie Proben auf Trockeneis, um eine Degradation zu verhindern.
- Vermeiden Sie wiederholte Frost-Tau-Zyklen.
- Für RNA-Proben in RNase-freiem Wasser oder Ethanol auflösen und bei -80 °C lagern.
- Kontaktieren Sie uns, wenn Ihr Proben-Typ nicht aufgeführt ist oder wenn Sie nur begrenztes Material haben; wir bieten maßgeschneiderte Lösungen für anspruchsvolle Projekte an.
Demonstrationsergebnisse
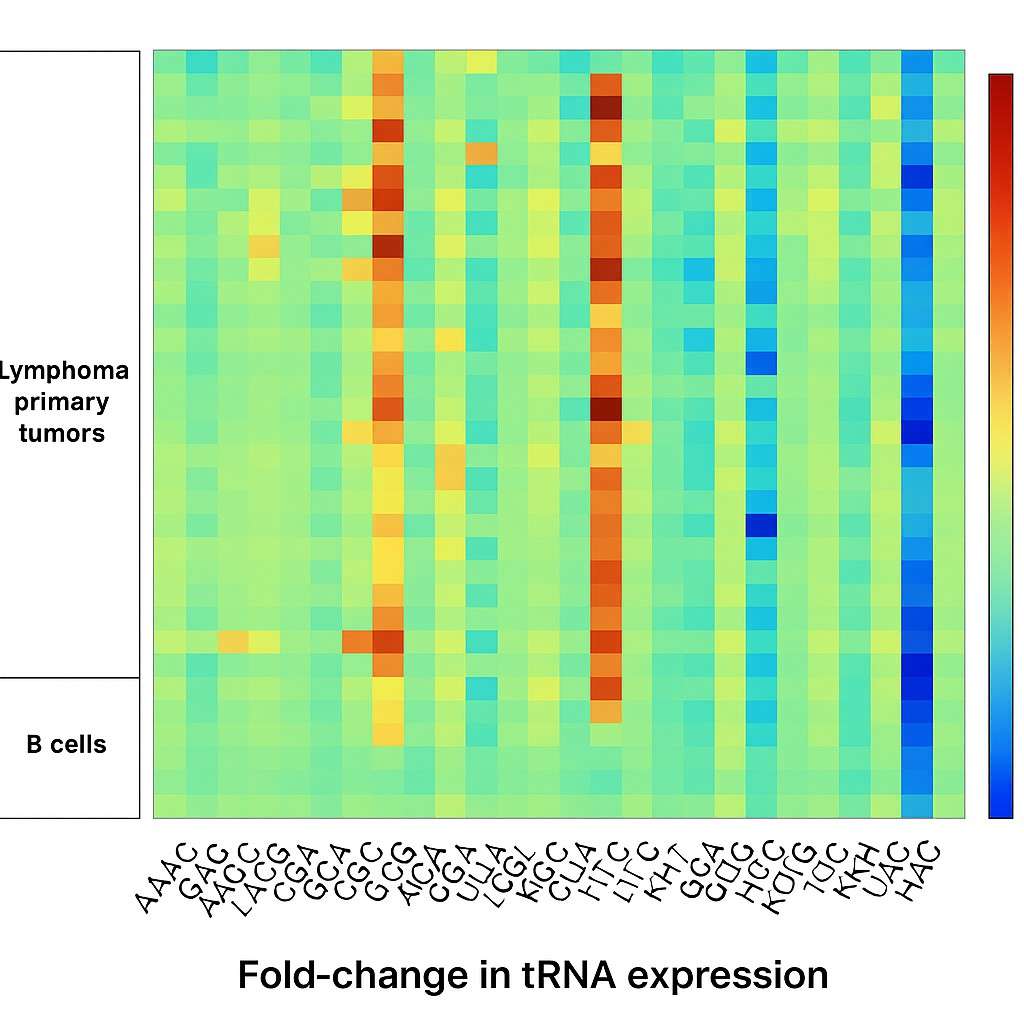
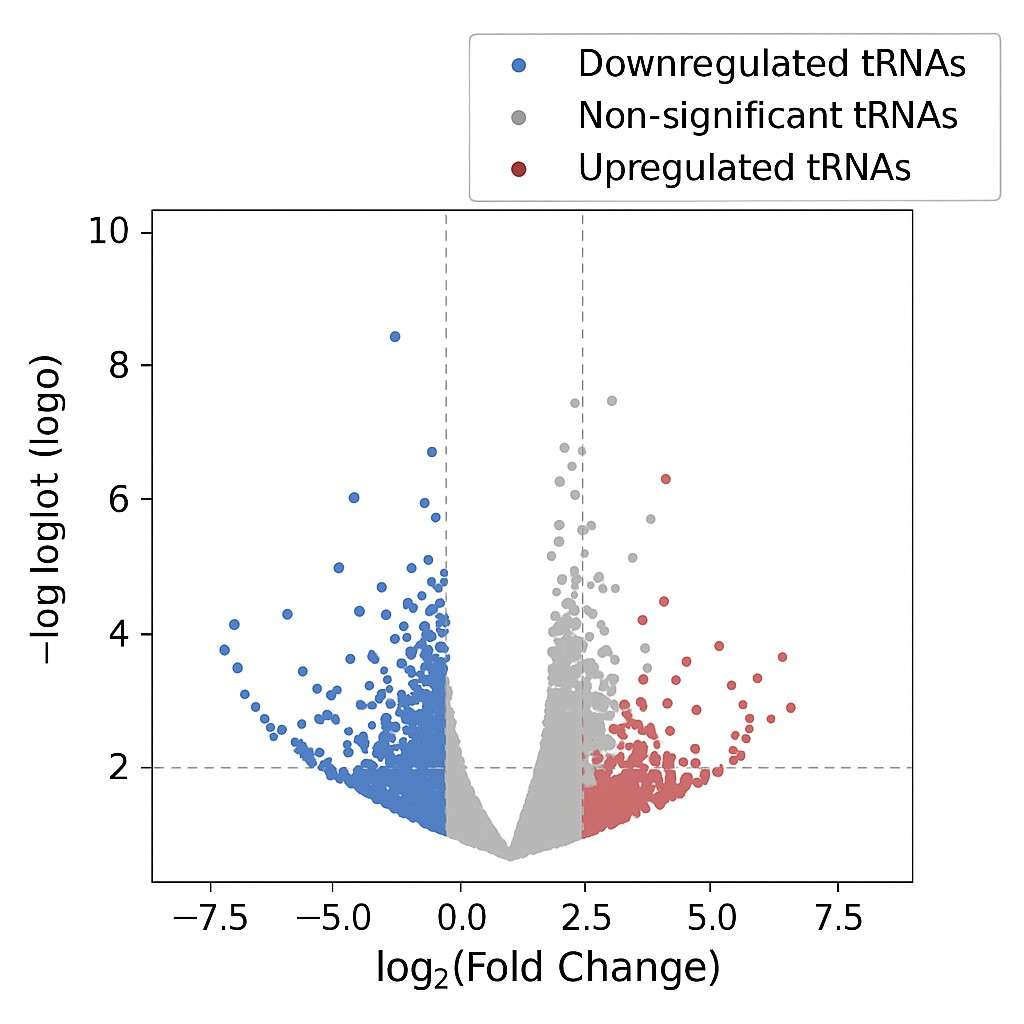
tRNA-Seq Häufig gestellte Fragen (FAQs)
Q1. Was ist tRNA-Sequenzierung und wie unterscheidet sie sich von anderen RNA-Seq-Methoden?
A1Die tRNA-Sequenzierung konzentriert sich speziell auf Transfer-RNA-Moleküle (~70–90 nt) und erfasst deren Überfluss, posttranskriptionale Modifikationenund Aminoacylierung (Ladestatus)Im Gegensatz zu standardmäßigen RNA-Seq, die sich auf mRNAs konzentriert und tRNA-Modifikationen sowie nuancierte Prozesse möglicherweise übersieht, unser tRNA-Sequenzierung mit Nanoporen und traditionell tRNA-Sequenzierungsmethoden sind optimiert, um die strukturierte und chemisch modifizierte Natur von tRNAs zu verarbeiten, was eine vollständige und modifikationsbewusste Profilierung ermöglicht.
Q2. Welche Sequenzierungsplattformen verwenden Sie?
A2CD Genomics verwendet sowohl Short-Read-Plattformen (z. B. Illumina) für die traditionelle tRNA-Sequenzierung als auch Drittgeneration-Nanoporen-Sequenzierung (z.B. Oxford Nanopore) für direkte, vollständige Länge tRNA-Sequenz und Modifikationsnachweis – ohne cDNA- oder PCR-Bias.
Q3. Welche Probenarten und Eingabemengen werden benötigt?
A3Wir akzeptieren Zellen, Gewebe, Körperflüssigkeiten (z. B. Blut, Liquor, Urin) und gereinigte Gesamtrna. Die Mindestmengen sind: 2×10⁶ Zellen, 50 mg Gewebe, 2–3 mL Blut/Serum/Plasma, 5 mL Liquor, 50 mL Urin oder ≥ 2 μg Gesamtrna (OD260/280 ≥ 1,8; OD260/230 ≥ 1,5). Proben sollten auf Trockeneis versendet und bei –80 °C gelagert werden, um die Integrität zu bewahren für tRNA-Sequenzierungsdienst. (Wie im Musteranforderungen Abschnitt.)
Q4. Welche Bearbeitungszeiten und Lieferungen kann ich erwarten?
A4Wir arbeiten effizient, um eine pünktliche Lieferung Ihrer Ergebnisse sicherzustellen. Kunden erhalten: interaktive bioinformatische Berichte, rohe Sequenzierungsdaten (FASTQ/BAM), quantitative Zählungen, Vulkan- und Heatmap-Visualisierungen, Modifikationszuordnungen, Aminoacylierungsprofiling und Grafiken in Publikationsqualität. Wir bestätigen einen detaillierten Zeitplan, wenn Sie Ihre Bestellung aufgeben.
Q5. Wie viele Proben oder Wiederholungen sollte ich einbeziehen?
A5Wir empfehlen mindestens drei biologische Replikate pro Gruppe um robuste differentiellen Ausdruck und statistische Analysen zu unterstützen. Während technische Replikate nicht erforderlich sind, bieten wir Bibliotheksmultiplexing die Kosten zu minimieren, ohne die Qualität zu opfern.
Q6. Welche Qualitätskontrollen sind vorhanden?
A6Wir führen während des gesamten Arbeitsablaufs strenge Qualitätskontrollen durch, indem wir:
- Proben-QC (Qubit, NanoDrop, Bioanalyzer)
- Bibliotheks-QC (Bioanalyzer, qPCR)
- Sequenzierungs-QC (plattform-spezifische Kontrollen)
- Daten-QC mit Tools wie FastQC
Alle QC-Ergebnisse sind in Ihrem Datenpaket enthalten.
Q7. Können Sie alle tRNA-Modifikationen erkennen und die Aminoacylierung messen?
A7Ja – wir erkennen eine Vielzahl von Modifikationen (z. B. Methylierung, Pseudouridylierung) durch den Vergleich von modifizierten und unmodifizierten Signalen in tRNA-Sequenzierung mit Nanoporen Daten. Geladene vs. ungeladene tRNAs sind über charakteristische Signale in speziellen Protokollen wie aa-tRNA-seq unterscheidbar.
Q8. Wie annotierst du tRNA-Sequenzen?
A8Sequenzen werden gegen unsere kuratierten ausgerichtet. tRNA-Sequenzdatenbankunter Verwendung von Werkzeugen wie tRNAscan-SE in Kombination mit modifikationsbewussten Algorithmen, um eine genaue Isoform- und Variantenannotation zu gewährleisten.
Q9. Ist eine Probe mit geringem Aufwand oder eine herausfordernde Probe verfügbar?
A9Definitiv. Wir unterstützen Low-Input-Workflows und maßgeschneiderte Bibliotheksvorbereitungen für begrenzte oder wertvolle Proben, indem wir Demethylierungs- und Adapterstrategien nutzen, um den Ertrag und die Datenqualität zu maximieren.
Q10. Wie kann ich anfangen?
A10Kontaktieren Sie unser Team, um die Projektziele, den Proben-Typ und den Forschungskontext zu besprechen. Wir werden einen maßgeschneiderten Plan vorschlagen, ein Angebot unterbreiten und Sie bei der Probenvorbereitung unterstützen. Sobald wir uns einig sind, senden Sie die Proben auf Trockeneis, und wir kümmern uns um den Rest – wir liefern hochwertige Daten und eine fachkundige Interpretation.
tRNA-Seq Fallstudien
Nano-tRNAseq: Quantitative tRNA-Häufigkeit und Modifikationsprofilierung
1. Hintergrund
Traditionelle Methoden zur Analyse von tRNA leiden unter erheblichen Verzerrungen aufgrund der reversen Transkription, der PCR-Amplifikation und der Unfähigkeit, tRNA-Modifikationen nachzuweisen. Da tRNAs etwa 13 Modifikationen pro Molekül tragen, die die Translation und Krankheiten beeinflussen, ist eine genaue Methode zur gleichzeitigen Quantifizierung von Häufigkeit und Modifikationen unerlässlich.
2. Methoden
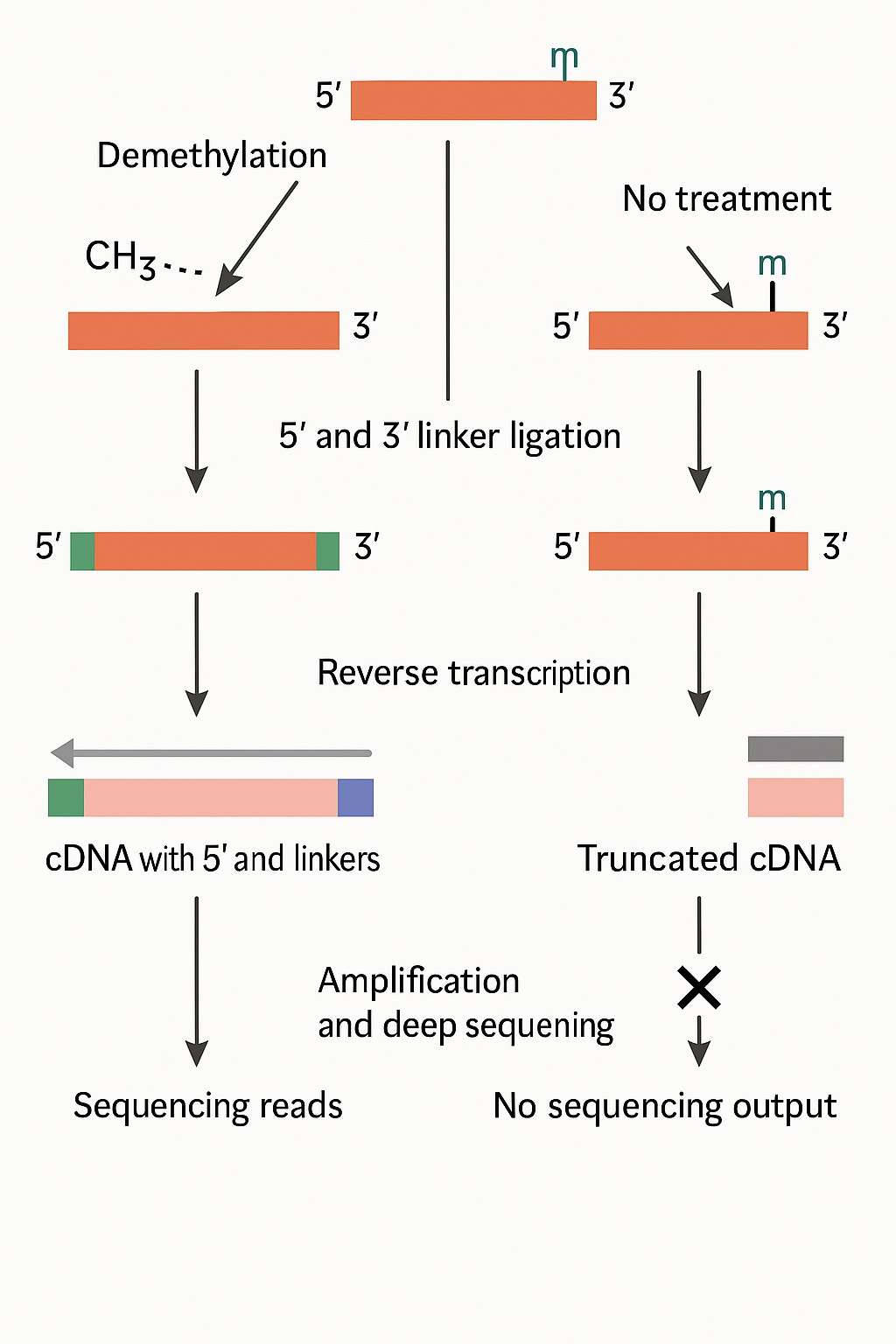
Die Autoren entwickelten Nano-tRNAseqein direktes RNA-Nanoporen-Sequenzierungsprotokoll, das Folgendes umfasst:
- Doppelte Ligierung von 5'- und 3'-Adaptern an reife tRNAs
- Demethylierung und Deacylierung zur Verbesserung der Adapterligierung
- Die erneute Verarbeitung von Roh-MinKNOW-Signalen zur Wiederherstellung von tRNA-Reads
- Vergleichende Sequenzierung von in vitro transkribierten (IVT) und nativen tRNAs aus S. cerevisiae
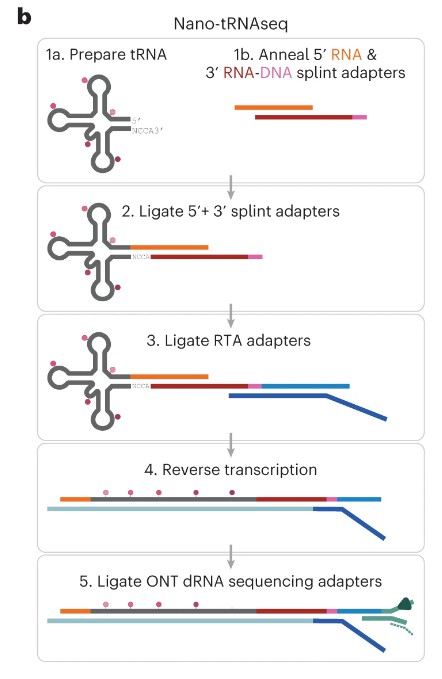 Abb. 1b mit dem Schemadiagramm zur Bibliotheksvorbereitung, das die Adapterligationen auf tRNA zeigt. Beschriften Sie dies unmittelbar unter dem Schemadiagramm.
Abb. 1b mit dem Schemadiagramm zur Bibliotheksvorbereitung, das die Adapterligationen auf tRNA zeigt. Beschriften Sie dies unmittelbar unter dem Schemadiagramm.
3. Ergebnisse
- 12-fache Erhöhung der tRNA-Lesungen mit benutzerdefinierten MinKNOW-Parametern im Vergleich zu den Standardparametern.
- Hohe Reproduzierbarkeit der Messungen von nativen tRNA (Spearmans ρ = 0,984).
- Genauigkeit der Quantifizierung der Abundanz im Vergleich zu Illumina-basierten Techniken (ρ = 0,93).
- Erkennung von Änderungsabhängigkeiten (z. B. Verlust von Ψ55, der m¹A58 und m⁵U54 beeinflusst) und CCA-Schwanz-Deadenylierung unter oxidativem Stress.
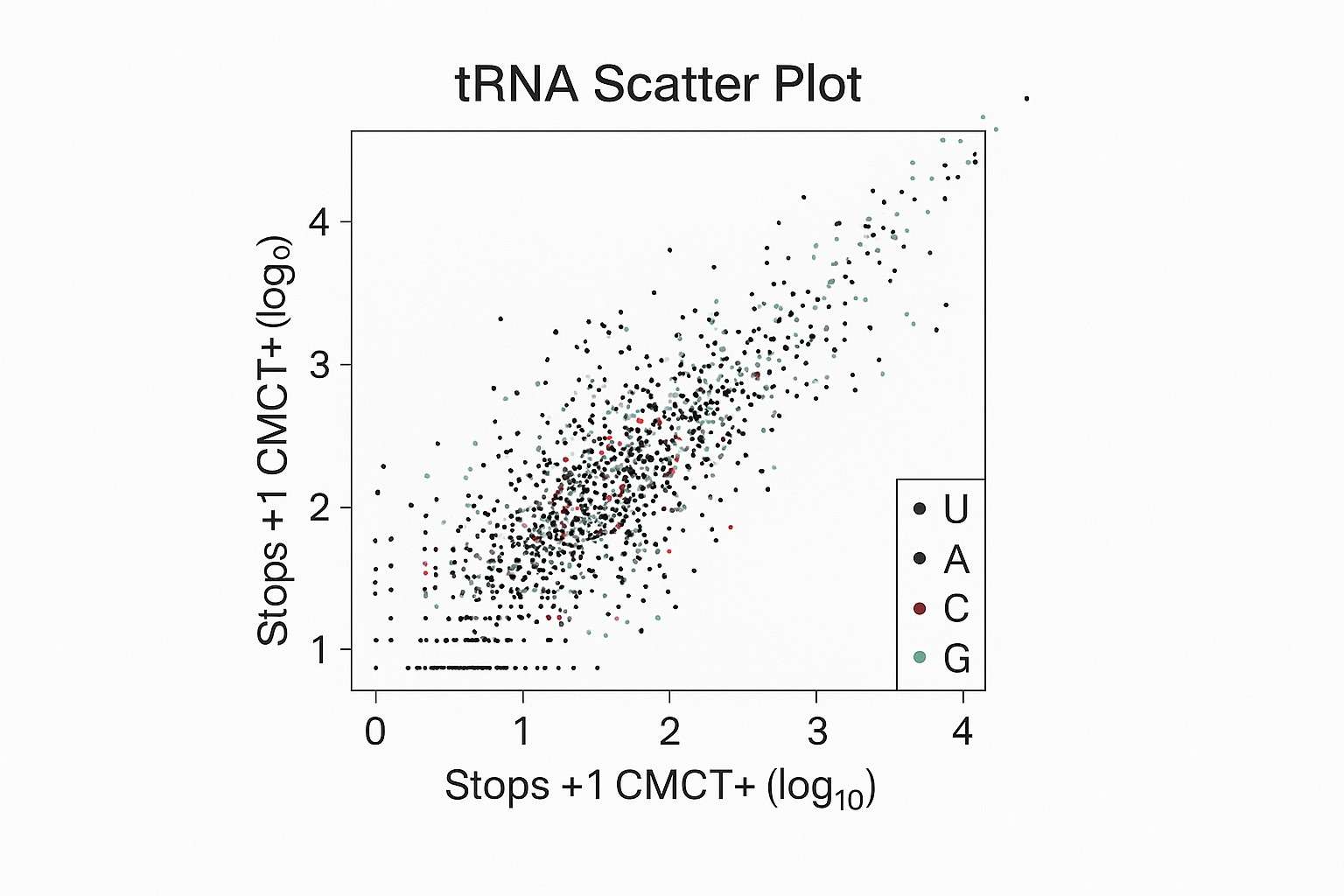
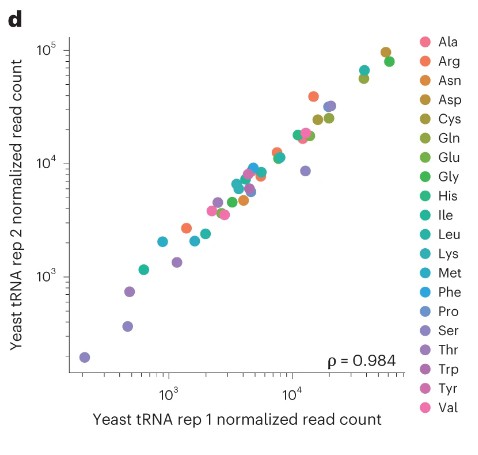 Abb. 1d Streudiagramm zur Darstellung der Reproduzierbarkeit
Abb. 1d Streudiagramm zur Darstellung der Reproduzierbarkeit
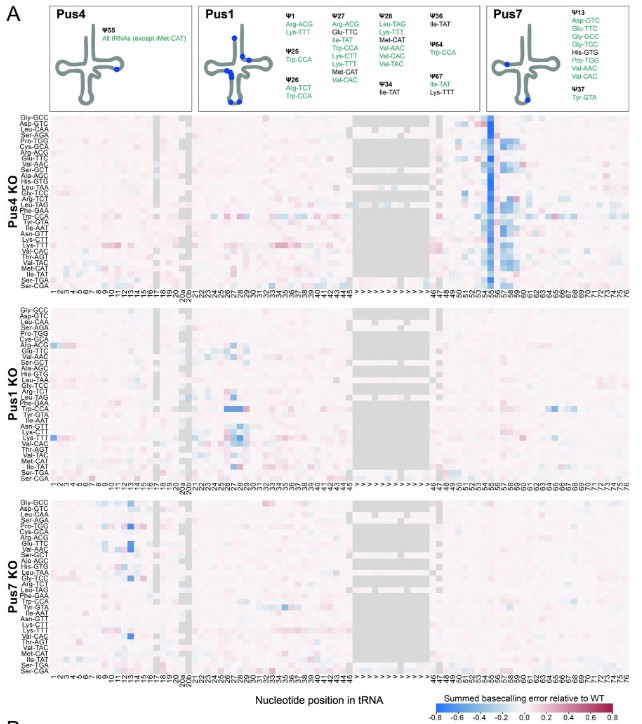 Abb. 9a Heatmap der Basisaufruf-Fehler im PUS4-Knockout.
Abb. 9a Heatmap der Basisaufruf-Fehler im PUS4-Knockout.
4. Schlussfolgerungen
Nano-tRNAseq bietet ein kosteneffektivein Hochdurchsatzansatz mit Einzelmolekülauflösung zur genauen Quantifizierung der tRNA-Häufigkeit und Modifikationszustände. Er überwindet die Verzerrungen traditioneller Methoden und etabliert einen Rahmen für das Studium des "tRNAome" bei Krankheiten, Stressreaktionen und der Entdeckung von Biomarkern.


