
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
mRNA-Sequenzierungsdienst
Was ist mRNA-Sequenzierung?
mRNA wird von protein-codierenden Genen transkribiert und dient als das intermediäre RNA-Molekül, das genetische Informationen von DNA zu Proteinen überträgt. In Eukaryoten wird DNA zunächst in prä-mRNA transkribiert, die eine Reihe von Verarbeitungsschritten durchläuft, um reife mRNA zu bilden. Die reife mRNA wird dann ins Zytoplasma transportiert und von Ribosomen in Proteine übersetzt. Die Reifung von mRNA umfasst drei Hauptverarbeitungsschritte: Spleißen, 5'-Capping und 3'-Polyadenylierung. Infolgedessen besitzen reife mRNA-Moleküle einen Poly(A)-Schwanz an ihrem 3'-Ende.
 Abb. 1. Was ist mRNA.
Abb. 1. Was ist mRNA.
mRNA-seq ist ein Hochdurchsatz-Sequenzierung Technologie, die zur Erkennung von mRNA-Expression und -Verarbeitung entwickelt wurde. Die Untersuchung der mRNA-Sequenzierung bei Eukaryoten konzentriert sich auf die Sammlung von mRNAs, die aus spezifischen Zellen oder Geweben unter bestimmten funktionalen Zuständen transkribiert werden. Sie kann verwendet werden, um differenzielle mRNA-Expression zu untersuchen, strukturelle Variationen zu erkennen und molekulare Marker zu identifizieren, die mit Krankheiten oder Eigenschaften assoziiert sind. Sie kann auch die Komplexität des Transkriptoms aufdecken, Gene und Transkriptstrukturen, variables Spleißen, RNA-Bearbeitung, polyadenylierte nicht-kodierende RNAs und neuartige Transkripte bestimmen. Derzeit wird mRNA-seq in Bereichen wie Grundlagenforschung, molekularer Züchtung, klinischer Diagnostik und Arzneimittelentwicklung.
mRNA-seq Technisches Prinzip
Für RNA mit Poly(A)-Schwänzen, wie mRNA und einigen lncRNAs, wird eine Anreicherung mit Oligo dT durchgeführt, die die polyadenylierten RNA einfängt, auch bekannt als PolyA-Seq. Die angereicherte mRNA/lncRNA wird dann fragmentiert, revers transkribiert, mit gemeinsamen Sequenzierungsadaptern ligiert und PCR-amplifiziert, um Sequenzierungsbibliotheken zu erzeugen. Die resultierenden Sequierungsdaten werden einer bioinformatischen Analyse unterzogen, um Informationen über Genexpression, alternatives Spleißen, RNA-Bearbeitung und andere Merkmale, die in der RNA vorhanden sind, zu erhalten.
 Abb. 2. Allgemeiner Arbeitsablauf von mRNA-seq.
Abb. 2. Allgemeiner Arbeitsablauf von mRNA-seq.
Vorteile unseres mRNA-Sequenzierungsdienstes
- Im Vergleich zu Genexpressions-Mikroarrays hat mRNA-Seq mehrere Vorteile in Transkriptomanalyse:
- Es hat einen breiteren dynamischen Bereich, der sowohl die Empfindlichkeit als auch die Genauigkeit bei der Messung von Faltänderungen in der Genexpression erhöht.
- Es kann sowohl bekannte Merkmale als auch neuartige Merkmale erfassen.
- Es kann breit auf verschiedene Arten angewendet werden.
- Neben der Berechnung der Genexpressionsniveaus kann es Sequenz- und Strukturvariationen in Transkripten erkennen.
- Eine erhöhte Sequenzierungstiefe ermöglicht einen breiteren dynamischen Erfassungsbereich, der die Identifizierung und Quantifizierung sowohl hochabundanter als auch niedrigabundanter Transkripte über einen Bereich von sechs Größenordnungen hinweg ermöglicht.
- Neben der Erkennung der Expressionsniveaus bekannter Transkripte kann es auch neuartige Transkripte entdecken.
- Wenn es auf Arten ohne Referenzgenom angewendet wird, kann es neue Gene durch de novo Assemblierung entdecken.
mRNA-Sequenzierungsdienst-Workflow
Für mRNA-Seq bietet unser Unternehmen experimentelle Dienstleistungen an, einschließlich, aber nicht beschränkt auf die Bibliothekskonstruktion und Sequenzierung basierend auf Geweben und Zelllinien sowie optimierte und maßgeschneiderte bioinformatische Analysen und Interpretationsdienste.

Dienstspezifikation
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategien
|
 |
Datenanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in der mRNA-Sequenzierung für Ihr Schreiben (Anpassung)
Bei CD Genomics bieten wir hochmoderne mRNA-Sequenzierungsdienste an, die auf verschiedene Forschungsbedürfnisse zugeschnitten sind. Unser Service umfasst eine strenge Qualitätskontrolle der Proben, eine fachkundige Bibliotheksvorbereitung, Hochdurchsatz-Sequenzierungund detaillierte Datenanalysen. Wir legen Wert darauf, präzise und umfassende Ergebnisse zu liefern, die mit den spezifischen Zielen Ihrer Forschung übereinstimmen. Kontaktieren Sie uns, um zu erfahren, wie unsere mRNA-Sequenzierungslösungen Ihre wissenschaftlichen Bestrebungen unterstützen und verbessern können.
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten aufgeführt:

Sequenzierungsqualitätsverteilung
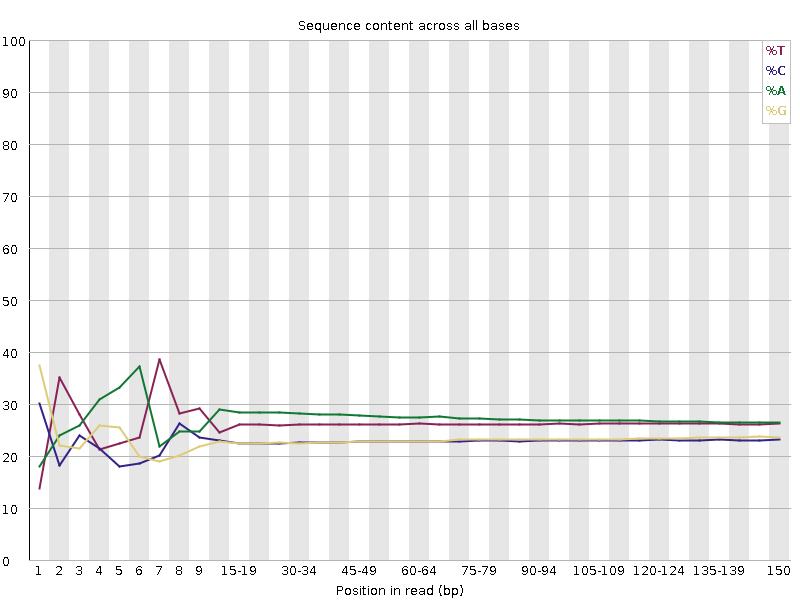
A/T/G/C-Verteilung
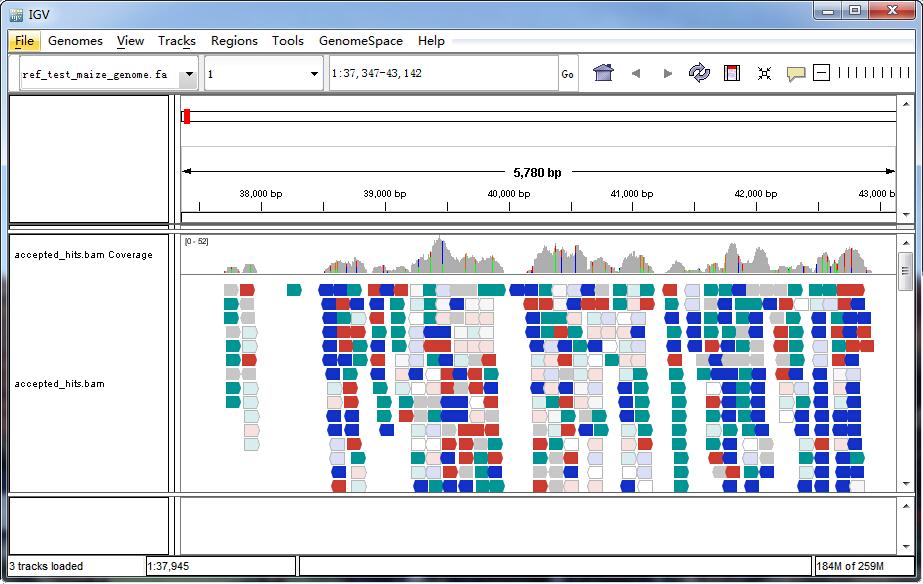
IGV-Browser-Oberfläche
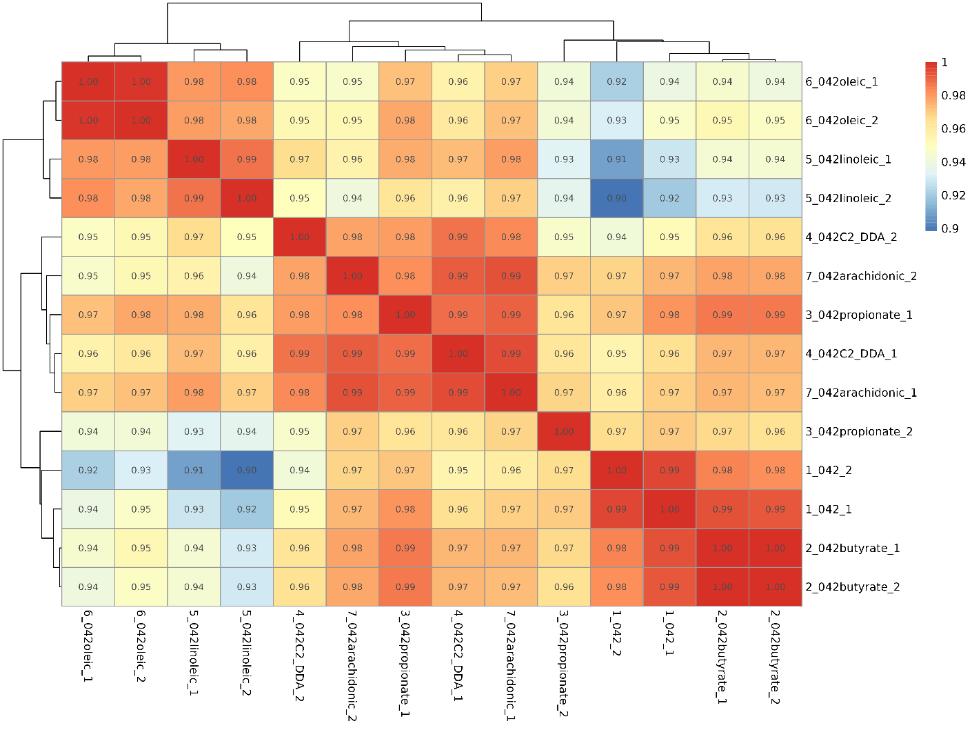
Korrelationsanalyse zwischen Proben
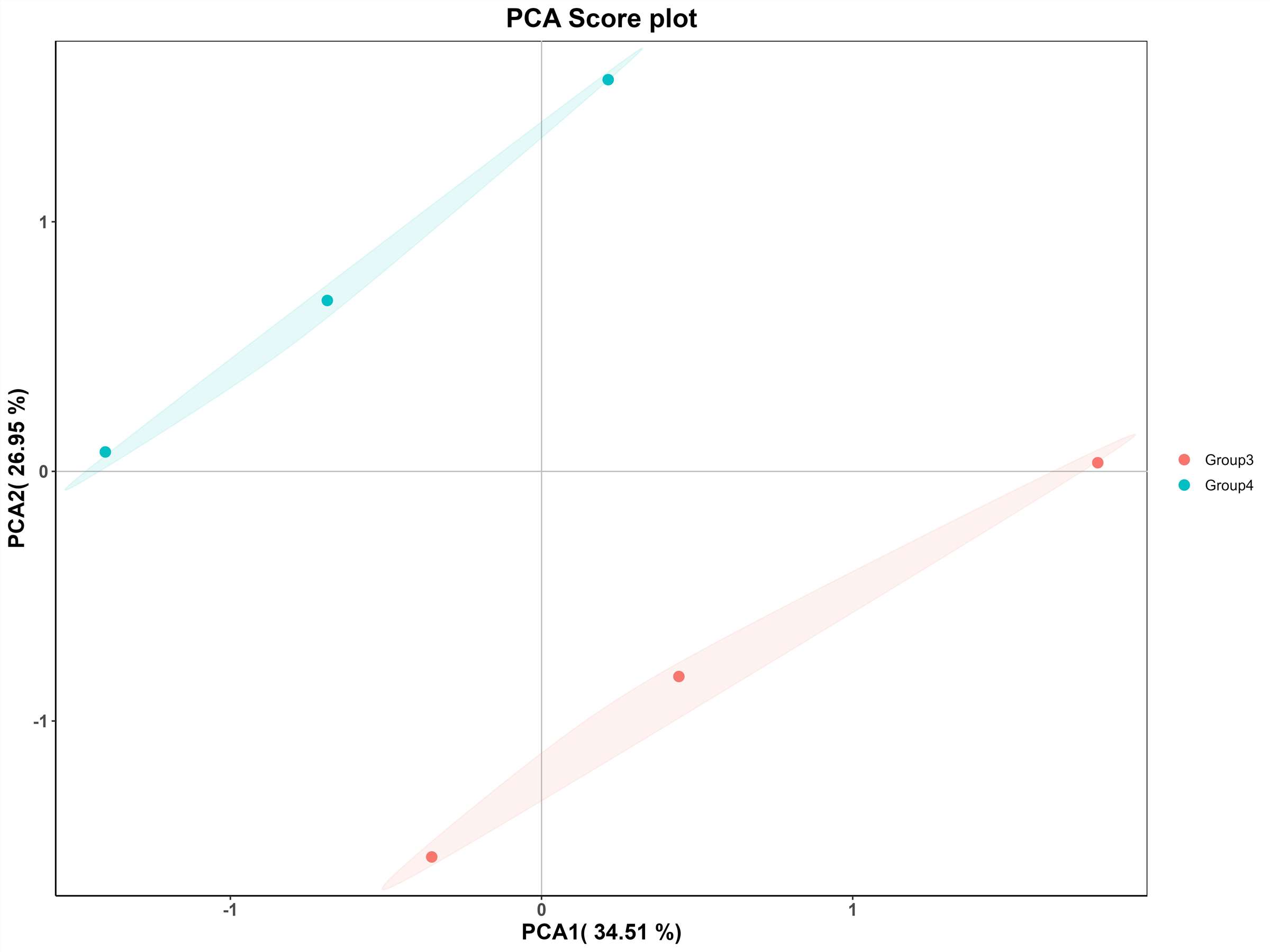
PCA-Score-Diagramm

Venn-Diagramm
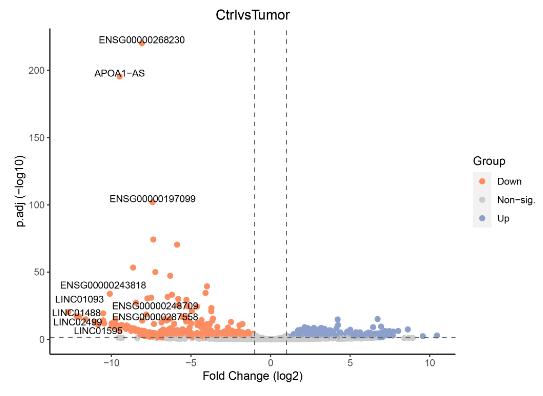
Volcano-Diagramm
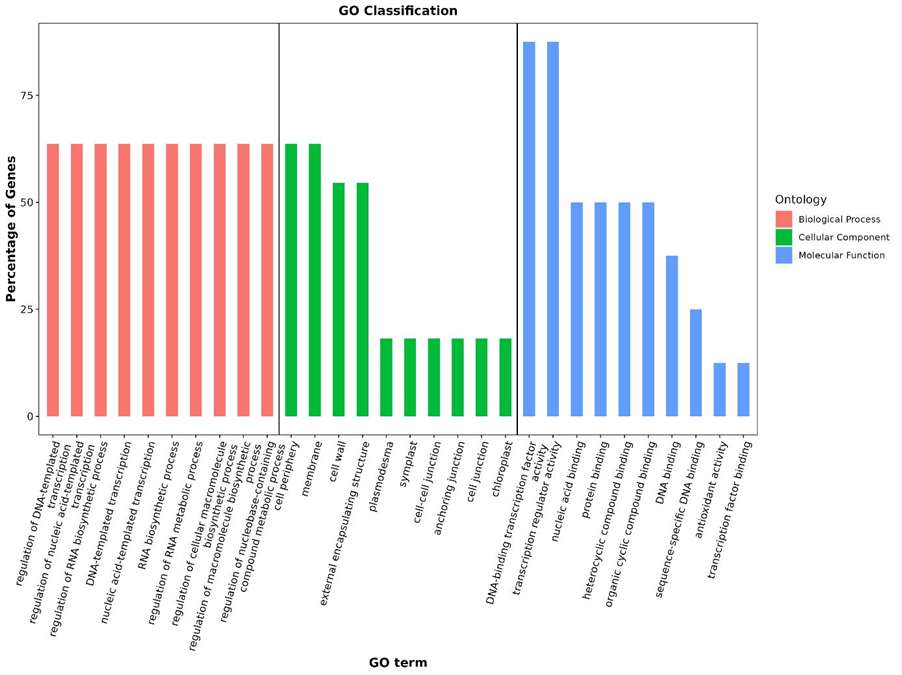
Statistik Ergebnisse der GO-Annotierung
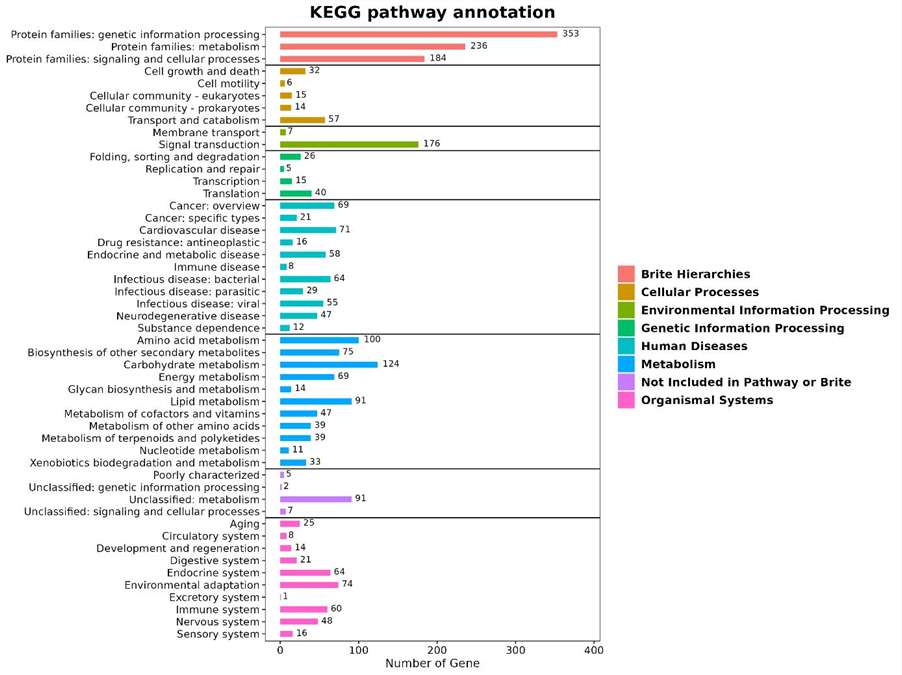
KEGG-Klassifikation
mRNA-Seq FAQs
Können alle Eukaryoten eine mRNA-Sequenzierung durchführen?
Eukaryotische mRNA enthält einen Poly-A-Schwanz, und die gängige Methode zur Anreicherung eukaryotischer mRNA besteht darin, mRNA aus totalem RNA mithilfe von magnetischen Perlen, die an Poly-T-Oligos gebunden sind, zu isolieren, gefolgt von der Bibliothekskonstruktion für das Sequenzieren. Daher können theoretisch alle Eukaryoten eine mRNA-Sequenzierung durchlaufen.
Warum erfordert die mRNA-Sequenzierung eine höhere RNA-Integrität als die gesamte Transkriptom-Sequenzierung?
Bei der Konstruktion von mRNA-Sequenzierungsbibliotheken werden RNA-Moleküle mit einem Poly-A-Schwanz mithilfe von magnetischen Perlen erfasst. Wenn die RNA-Integrität schlecht ist und die mRNA teilweise abgebaut wird, was zu einem Verlust des Poly-A-Schwanzes führt, ist die erfasste mRNA aus Proben mit geringerer Integrität reduziert, was zu einer geringeren Genabdeckung führt. Daher ist ein höheres Maß an RNA-Integrität erforderlich.
3. Kann mRNA-Seq nicht-kodierende RNA nachweisen?
mRNA-seq erfasst hauptsächlich RNA-Moleküle mit einem Poly(A)-Schwanz; daher können theoretisch alle in der Zelle exprimierten Moleküle mit einem Poly(A)-Schwanz nachgewiesen werden. Dazu gehören mRNA und lange nicht-kodierende RNAs mit Poly(A)-Schwänzen.
4. Was ist der Unterschied zwischen totalem RNA-Seq und mRNA-Seq?
Gesamt-RNA-Seq, oder die gesamte Transkriptom-Sequenzierung, ist eine umfassende Methode, die die Sequenzierung aller RNA-Spezies nach der Entfernung von ribosomaler RNA (rRNA) umfasst, einschließlich kodierender und nicht-kodierender RNAs. Dazu gehören RNA wie rRNA, prä-mRNA, messenger RNA (mRNA) und eine Vielzahl von nicht-kodierenden RNAs (ncRNA) wie Transfer-RNA (tRNA), Mikro-RNA (miRNA) und lange nicht-kodierende RNA (lncRNA). Im Gegensatz dazu zielt mRNA-Seq speziell auf kodierende Regionen ab, indem es polyadenylierte (poly(A)) RNA anreichert, was es zu einem kostengünstigeren und effizienteren Ansatz für Studien macht, die sich hauptsächlich auf die mRNA-Analyse konzentrieren.
5. Sollten Sie sich für mRNA-Sequenzierung oder totale RNA-Sequenzierung entscheiden?
ist die mRNA-Seq-Methodik die optimale Wahl für Forschungsprojekte, die eukaryotische Organismen betreffen und ein primäres Interesse an kodierenden Regionen haben. Der mRNA-Seq-Ansatz reichert mRNA durch Poly(A)-Selektion an, was die Sequenzierungskosten und die Komplexität reduziert und besonders für Proben mit begrenztem Ausgangsmaterial geeignet ist. Wenn hingegen eine umfassende Analyse erforderlich ist, die alle RNA, einschließlich kodierender und nicht-kodierender RNA-Moleküle, umfasst, total RNA-Seq ist die bevorzugte Option, trotz der höheren Kosten und des größeren Bedarfs an Sequenzierungsdaten. Die Auswahl zwischen den Methoden sollte auf spezifischen experimentellen Zielen, biologischen Fragestellungen, Probenarten und Budgetbeschränkungen basieren.
mRNA-Seq Fallstudien
Molekularer Mechanismus der Differenzierung der Cadmiumtoleranz in Lentinula edodes wie durch mRNA- und miRNA-Analysen offenbart
Zeitschrift: Zeitschrift für gefährliche Materialien
Impactfaktor: 14,224
Veröffentlicht: 15. Oktober 2022
Hintergrund
Cadmium (Cd) ist ein weit verbreitetes giftiges Schwermetall, das in der Lage ist, mehrere Krankheiten im menschlichen Körper zu induzieren, wie Brustkrebs und Nierenkrebs. Mit der raschen Entwicklung industrieller und landwirtschaftlicher Aktivitäten ist die Cadmiumkontamination zu einem ernsthaften globalen Problem geworden. Daher ist es unerlässlich, Cd-kontaminierte Gebiete zu sanieren. Die fungale Sanierung ist eine neuartige und wichtige Wiederherstellungstechnik, doch die molekularen Mechanismen, die den Reaktionen von Pilzen auf Schwermetallstress zugrunde liegen, sind nach wie vor unklar. Diese Studie führte durch Transkriptom-Sequenzierung Um die differenzielle mRNA- und miRNA-Expression zwischen Cd-toleranten (YS119) und Cd-sensiblen (YS45) Pilzen zu analysieren. Die Ergebnisse liefern eine theoretische Grundlage für die weitere Erforschung der molekularen Mechanismen der Cadmiumtoleranz in Pilzen und helfen, die potenziellen Rollen von miRNAs in den Reaktionen von Pilzen auf Schwermetallstress zu erhellen.
Materialien & Methoden
- Cd-tolerante Pilz YS119
- Cd-sensitives Pilz YS45
- RNA-Extraktion
- mRNA-Sequenzierung
- miRNA-seq
- PCA und Korrelationsanalyse
- DEGs-Analyse
- miRNA-Zielgenvorhersage
- GO- und KEGG-Analyse
- Integrierte Analyse von mRNAs und miRNAs
Ergebnisse
1. mRNA-Sequenzierungsanalyse
Insgesamt wurden 577.586.098 Rohdaten aus allen Proben gewonnen, was nach der Filterung zu 577.165.672 sauberen Daten führte. Die Pearson-Korrelationskoeffizienten und die Hauptkomponentenanalyse (PCA) zeigten eine hohe Reproduzierbarkeit zwischen den biologischen Replikaten der Proben, was eine weitergehende Analyse ermöglichte (Abbildung 1A, B).
 Abbildung 1 Pearson-Korrelationskoeffizient und PCA-Analyse.
Abbildung 1 Pearson-Korrelationskoeffizient und PCA-Analyse.
Unter Cd-Stress zeigte YS45 eine stärkere Reaktion im Vergleich zu YS119, mit 2036 differentially expressed genes (DEGs) in YS45 (1034 hochreguliert, 1002 herunterreguliert) im Vergleich zu 370 DEGs in YS119 (239 hochreguliert, 131 herunterreguliert). GO- und KEGG-Analysen zeigten, dass beide Pilzsorten 170 gemeinsam hochregulierte DEGs hatten, die in 14 GO-Kategorien und einem KEGG-Weg angereichert waren, sowie 107 gemeinsam herunterregulierte DEGs, die in 12 GO-Kategorien und einem KEGG-Weg angereichert waren, was darauf hinweist, dass Gene, die an der Proteinstabilität, der Ionenbindung und der Redoxaktivität beteiligt sind, eine Rolle in der Cd-Stressreaktion spielen. Konkret hatte YS45 863 einzigartig hochregulierte Gene, die in 20 GO-Kategorien und 12 KEGG-Wegen angereichert waren, während YS119 64 einzigartig hochregulierte Gene hatte, die in 11 GO-Kategorien und einem KEGG-Weg angereichert waren.
Zusätzlich wies YS45 890 einzigartig herunterregulierte Gene auf, die in 22 GO-Kategorien und 8 KEGG-Wegen angereichert waren, während YS119 23 einzigartig herunterregulierte Gene in 7 GO-Kategorien ohne KEGG-Weg-Anreicherung hatte. Dies deutet darauf hin, dass die schwere Unterdrückung von Genen, die mit der Umgestaltung der Zellwand, DNA-Reparatur, Zuckerstoffwechsel und Proteolyse unter Cd-Stress in Verbindung stehen, die Cd-Toleranz von YS45 verringern könnte, wobei die differentielle Expression von Genen, die an der Transkriptionsregulation, dem Transport, dem Lipidstoffwechsel, der Metallbindung, dem Glutathionstoffwechsel und der Redox-Homöostase beteiligt sind, zu den Toleranzunterschieden zwischen den beiden Sorten beiträgt.
 Abbildung 2 DEGs-Analyse.
Abbildung 2 DEGs-Analyse.
2. Integrierte Analyse von mRNA und miRNA
miRNAs regulieren die Genexpression, indem sie ihre Ziel-mRNAs abbauen oder hemmen. In YS45 sind unter 1620 DE miRNA-Zielen 216 DEGs, wobei 111 entgegengesetzte Ausdruckstrends zu den miRNAs zeigen (Abbildung 3A, B). Gene, die potenziell negativ von diesen miRNAs reguliert werden, umfassen solche, die Transkriptionsfaktoren, Transportproteine, Cytochrom P450s, DNA-Reparaturproteine wie MutL, Proteasen, Glutathion-S-Transferasen und carbohydrate-active enzymes (CAZymes) kodieren. In YS119 sind von 525 DE miRNA-Zielen nur 14 DEGs, wobei 3 entgegengesetzte Ausdruckstrends zeigen (Abbildung 3C, D), die Glycosidase, Zellulose-Wachstumsprotein und Retinol-Dehydrogenase kodieren.
Diese Ergebnisse deuten darauf hin, dass miRNAs die Expression der meisten Zielgene in Pilzen möglicherweise nicht signifikant beeinflussen. Der mRNA-Abbau könnte nicht der primäre regulatorische Mechanismus sein; stattdessen könnten die translationalen Hemmungen und der Wettbewerb mit endogenen RNAs um die Bindung von miRNAs eine Schlüsselrolle spielen.
 Abbildung 3 Integrierte Analyse von mRNA und miRNA.
Abbildung 3 Integrierte Analyse von mRNA und miRNA.
Fazit
Diese Studie untersucht die bemerkenswerten molekularen Mechanismen, die den Unterschieden in der Cadmium (Cd)-Toleranz zwischen zwei Pilzsorten, YS45 und YS119, zugrunde liegen. Unter Cd-Stress zeigte YS45 eine größere Proteinaggregation im Vergleich zu YS119, was die normalen zellulären Aktivitäten beeinträchtigte und die Cd-Toleranz des Pilzes verringerte. Vergleichende Analysen von mRNA und miRNA zeigten die Beteiligung von Genen oder miRNAs, die mit Zellwandumbau, Transport, Metallchelation, Redox-Homöostase, Signaltransduktion, transkriptioneller Regulation, Lipid- und Kohlenhydratstoffwechsel, Proteinabbau, DNA-Reparatur und Zellzyklusregulation in der Modulation der Cd-Toleranz assoziiert sind. Darüber hinaus bestätigte die Überexpression des LeAmy-Gens seine Rolle bei der Verbesserung der Cd-Toleranz in Pilzen. Die Ergebnisse legen eine theoretische Grundlage für ein tieferes Verständnis der molekularen Mechanismen hinter den Unterschieden in der Cd-Toleranz bei Pilzen und unterstützen die Entwicklung von schwermetalltoleranten Pilzkulturen.
Referenz:
- Shen N, Xu C, Zhang J, et al. Molekulare Mechanismen der Differenzierung der Cadmiumtoleranz in Lentinula edodes wie durch mRNA- und miRNA-Analysen offenbart. Zeitschrift für gefährliche Materialien, 2022, 440: 129841.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Menschliche unreife Zahnmark-Stammzellen haben sich nicht in ein bereits bestehendes menschliches Lungenadenokarzinom eingepflanzt.
Zeitschrift: Fallberichte in der Onkologie
Jahr: 2022
Zirkuläre DNA-Tumorviren erzeugen zirkuläre RNAs.
Zeitschrift: Mitteilungen der Nationalen Akademie der Wissenschaften
Jahr: 2018
Wiederholte Immunisierung mit ATRA-haltigem liposomalem Adjuvans transdifferenziert Th17-Zellen zu einem Tr1-ähnlichen Phänotyp.
Zeitschrift: Zeitschrift für Autoimmunität
Jahr: 2024
Die Rolle der Histonvariante H2A.Z.1 bei Gedächtnis, Transkription und alternativer Spleißung wird durch Lysinmodifikation vermittelt.
Zeitschrift: Neuropsychopharmakologie
Jahr: 2024
FAK-Verlust reduziert die ERK-Phosphorylierung, die durch BRAFV600E induziert wird, um die intestinale Stammzelligkeit und die Tumorbildung im Blinddarm zu fördern.
Journal: Elife
Jahr: 2023
Identifizierung von zirkulären RNAs, die die Proliferation von Kardiomyozyten in neugeborenen Schweineherzen regulieren
Journal: JCI Insight
Jahr: 2024
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


