
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
ddRAD-seq
Mit der umfangreichen Expertise von CD Genomics in Next-Generation-Sequenzierung (NGS)Wir freuen uns, nun ddRADseq-Dienste (Double Digest Restriction-site Associated DNA) für umfassende genomweite SNP-Entdeckung anzubieten, selbst in Abwesenheit vorheriger genomischer Sequenzinformationen. ddRADseq ermöglicht die Sequenzierung von genomweiten Daten aus Nicht-Modellarten und fördert die kosteneffiziente Entwicklung umfangreicher Datensätze, die eine breite taxonomische und geografische Stichprobe umfassen.
Was ist ddRAD-Sequenzierung?
ddRADseq hat bei Molekularökologen beträchtliche Popularität erlangt, um neuartige SNP-Marker unter Verwendung von NGS-Plattformen zu entwickeln. Diese Methode nutzt die Schnittstellen-Spezifität von Restriktionsendonuklease-Enzymen, um Bibliotheksfragmente aus verschiedenen genomischen Regionen zu erzeugen, die bei Individuen derselben Art konserviert sind. Diese Konservierung ermöglicht die Sequenzierung und den vergleichenden Vergleich identischer genomischer Regionen bei verschiedenen Individuen. Folglich erleichtert ddRADseq die schnelle und effiziente Entwicklung einer beträchtlichen Anzahl genetischer Marker.
Was ist das Prinzip der ddRAD-Sequenzierung?
ddRADseq ist eine verbesserte Variante des traditionellen RAD-Sequenzierung Protokoll, das speziell für die SNP-Entdeckung und GenotypisierungStatt Fragment-Scheren integriert ddRAD-seq eine sekundäre Restriktionsverdauung, wodurch sowohl die Anpassungsfähigkeit als auch die Genauigkeit des Größenwahl-Schrittes verbessert werden. Kurz gesagt, beginnt der Prozess mit dem Verdau von genomischer DNA unter Verwendung eines Restriktionsenzym, gefolgt von der Ligation eines barcodierten P1-Adapters an die resultierenden Fragmente. Diese adapter-ligierten Fragmente aus verschiedenen Proben werden anschließend zusammengeführt, und ein zweites Restriktionsenzym wird für einen weiteren Verdau angewendet. Die verdauten Fragmente werden dann einer Größenwahl und Reinigung unterzogen. Danach werden P2-Primer ligiert und die Fragmente amplifiziert. Schließlich werden die Sequenzdaten analysiert, um genetische Variationen innerhalb der interessierenden Proben zu bewerten und zu erfassen.
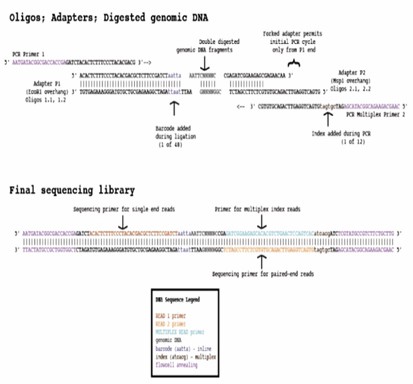
Was sind die Vorteile des ddRAD-Sequenzierungsdienstes?
- Wiederverwendbarkeit von Proben: Mehrere Proben können wiederverwendet werden, um die Ressourceneffizienz zu maximieren.
- Effizienter Arbeitsablauf: Der Prozess der Bibliotheksvorbereitung ist einfacher und schneller, was das gesamte experimentelle Verfahren optimiert.
- Konsistente Fragmentlängen: Gewährleistet einheitliche Fragmentlängen über Proben an identischen Verdauungsstellen, was die Genauigkeit verbessert.
- SNP-Dichte-Flexibilität: Bietet Flexibilität in der SNP-Dichte und ermöglicht eine Anpassung basierend auf spezifischen Forschungsbedürfnissen.
- Hohe Sequenzabdeckung: Erreicht eine hohe Sequenzabdeckung, die die Datenzuverlässigkeit erhöht.
- Kosten-Effektivität: Senkt die experimentellen Kosten bei gleichzeitiger Erhaltung robuster Datenoutputs.
- Kein Referenzgenom erforderlich: Ermöglicht die SNP-Entdeckung ohne die Notwendigkeit eines Referenzgenoms.
- Fortschrittliche Multi-Technologie-Plattform: Bietet zuverlässige Daten Ergebnisse, die sowohl den Anforderungen an Zuverlässigkeit als auch an Kosteneffizienz entsprechen.
- Technischer Support: Umfassende Unterstützungsdienste decken alle Projektbedürfnisse ab und gewährleisten die Kundenzufriedenheit.
- Optimierter experimenteller Prozess: Vereinfacht den experimentellen Prozess und gewährleistet eine schnelle Bearbeitung. Multi-Locus-Projekte können in multiplexen Tests durchgeführt werden, wobei die Typisierung in einem einzigen Röhrchen abgeschlossen wird.
- Echtzeit-Projektupdates: Bietet Echtzeit-Updates zum Projektfortschritt, sodass Kunden jeden Schritt klar verfolgen können.
- Angepasste Protokolle: Flexibilität, experimentelle Protokolle an projektspezifische Bedürfnisse anzupassen, mit fachkundiger Anpassung für optimale Ergebnisse.
Anwendungen der ddRAD-Sequenzierung
- Populationsgenetik
- Genomweite Assoziationsstudien (GWAS)
- Naturschutzbiologie
- Evolutionsbiologie
- Genomische Kartierung
- Zuchtprogramme
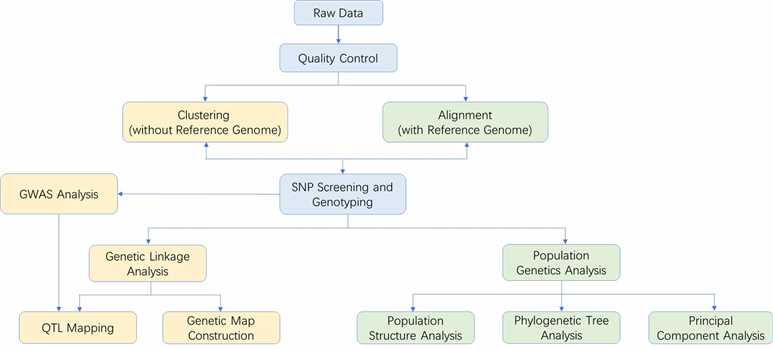
ddRAD-Sequenzierungs-Workflow
Der Arbeitsablauf der ddRAD-Sequenzierung bei CD Genomics umfasst mehrere wichtige Schritte:

Dienstspezifikationen
Beispielanforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in ddRAD-seq für dein Schreiben (Anpassung)
CD Genomics bietet einen umfassenden ddRAD-seq-Service an, der alles von den ersten DNA-Qualitätsprüfungen bis hin zur detaillierten SNP-Analyse abdeckt. Wir bieten auch maßgeschneiderte Lösungen an, die auf Ihre spezifischen Projektbedürfnisse zugeschnitten sind. Für weitere Informationen oder um Ihre Anforderungen zu besprechen, kontaktieren Sie bitte unser Team.
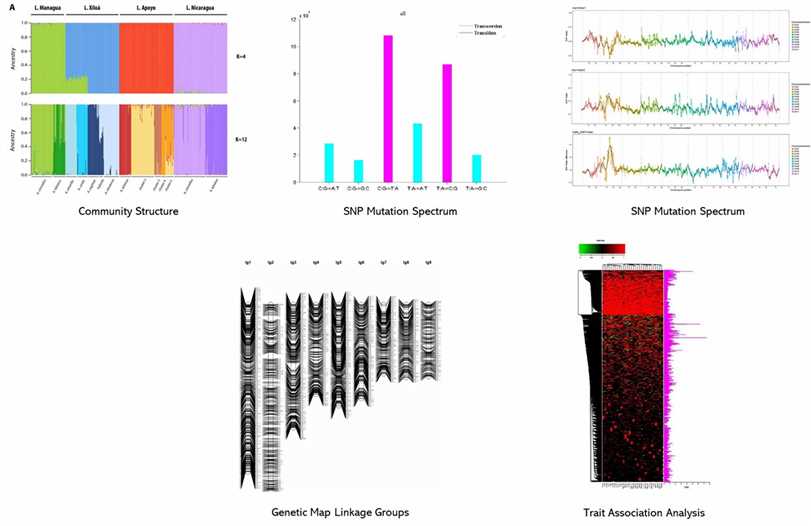
Demonstrationsergebnisse
Teilweise Ergebnisse sind unten angezeigt:
ddRAD-seq häufig gestellte Fragen (FAQs)
1. Wie lange dauert es, Ergebnisse von ddRAD-seq zu erhalten?
Die typische Bearbeitungszeit für ddRAD-seq-Ergebnisse variiert je nach Projektgröße und -komplexität, liegt jedoch normalerweise zwischen 4 und 8 Wochen.
2. Können wir den ddRAD-seq-Service an spezifische Bedürfnisse anpassen?
Ja, wir bieten maßgeschneiderte Lösungen, um spezifische Projektanforderungen zu erfüllen, einschließlich individueller Protokollanpassungen und Datenanalyseoptionen.
3. Welche Arten von Proben sind für ddRAD-seq geeignet?
ddRAD-seq kann auf verschiedene Probenarten angewendet werden, einschließlich Blut, Gewebe und anderen genomischen DNA-Quellen von Pflanzen, Tieren und Mikroorganismen.
4. Kann ddRAD-seq ohne ein Referenzgenom verwendet werden?
Ja, ddRAD-seq benötigt kein Referenzgenom, was es für Arten geeignet macht, für die keine Referenzgenome verfügbar sind.
5. Wie wählt man die geeignete Technologie zur reduzierten Genomdarstellung aus?
Bei der Auswahl der geeigneten vereinfachten Genomik-Technologie sollten Sie die folgenden vier Faktoren berücksichtigen, um Ihre Forschungsstrategie zu entwerfen:
Referenzgenom
- Mit Referenzgenom: Die Verwendung eines Referenzgenoms hilft, Fehler aufgrund homologer oder repetitiver Sequenzen zu reduzieren, erleichtert die LD-Analyse, die Selektionsanalyse und GWAS. Dies ist geeignet für konventionelle RAD-Sequenzierung und PE151-Sequenzierung.
- Ohne Referenzgenom: ddRAD-seq wird empfohlen.
Sequenzierungsstrategie
- Double Digest: Bei kurzen Fragmenten kann das Pair-End (PE) Sequencing zu erheblichem Adapterüberlapp führen und ist für lange Leseweiten nicht ideal; für kurze Fragmente wird das Single-End (SE) Sequencing empfohlen.
- Lange Fragmente: Lange Reads können mehr Varianteninformationen erkennen, erfordern jedoch eine ausreichende Abdeckung.
- Kurze Lesevorgänge: Verbessert die Sequenzierungstiefe pro Restriktionsstelle und erhöht die Genauigkeit der SNP-Erkennung.
- Nicht-Referenzarten: SE-Sequenzierung wird empfohlen, um Datenverschwendung zu vermeiden.
Anzahl der Marker
- Hohe Marker-Dichte: Konventionelles RAD-Sequencing eignet sich für Analysen, die eine hohe Dichte an Markern erfordern.
- Komplexe Genome und große Stichprobengrößen: GBS Die Sequenzierung ist ideal für komplexe Genome und große Probenmengen.
PCR-Amplifikationsartefakte
- PCR-Bias: Kann dazu führen, dass heterozygote Stellen fälschlicherweise als homozygot identifiziert werden oder Fehler einführen. Konventionelles RAD-Sequencing kann PCR-Duplikate mithilfe von Fragmentsequenzinformationen entfernen, während GBS und ddRAD-seq dies nicht können.
ddRAD-seq Fallstudien
ddRAD-Sequenzierung-basierte Genotypisierung zur Analyse der Populationsstruktur in kultiviertem Tomaten bietet neue Einblicke in die genomische Vielfalt des mediterranen 'da serbo'-Typs mit langer Haltbarkeit von Genmaterial. Horticulture Research
Zeitschrift: Gartenbau-Forschung
Impact-Faktor: 5,404
Veröffentlicht: 01. September 2020
Hintergrund
TomateSolanum lycopersicum L..) steht als eine wichtige wirtschaftliche Gemüsepflanze mit globalem Anbau. Die Anfänge der Tomatendomestikation lassen sich auf die Andenregion in Südamerika zurückverfolgen, von wo aus sie sich über die Amerikas verbreitete. Im 16. Jahrhundert wurden Tomaten nach Europa eingeführt, wobei Spanien und Italien als wichtige Eingangspunkte dienten. Diese Einführung löste weitere Domestikationsbemühungen aus, die zur Entstehung einer erheblichen lokalen genetischen Vielfalt beitrugen. Folglich wird das Mittelmeergebiet in Europa als sekundäres Zentrum für die Diversifizierung von Tomaten anerkannt. Im Rahmen dieser Untersuchung nutzten die Autoren die ddRAD-seq-Technologie, um die genetische Vielfalt innerhalb verschiedener Tomatensorten systematisch zu erforschen.
Materialien & Methoden
Probenvorbereitung
- Tomate
- DNA-Extraktion
Methode
- RAD-seq Genotypisierung
- HiSeq2500-Gerät
- SNP-Aufruf
- Filterung und Marker-Klassifizierung
- Bevölkerungsstruktur
- Genetische Vielfalt
- Phänotypische Analyse
Ergebnisse
In dieser Untersuchung wurde die genetische Vielfalt von 288 Tomatenspezimen untersucht, darunter 152 Proben der Langzeitlagerungssorte (LSL) 'da serbo', die hauptsächlich aus Italien und Spanien stammen, sowie andere gängige Sorten aus verschiedenen Ländern. Neben dem LSL-Merkmal zeigen 'da serbo'-Sorten auch Eigenschaften der Stressresistenz. Um nicht-parametrisches hierarchisches Clustering durchzuführen und die ancestrale Populationsstruktur zu modellieren, fand die Studie 32.799 hochqualitative Einzel-Nukleotid-Polymorphismen (SNPs). Sechs distinct genetische Subpopulationen wurden abgegrenzt, die die Mehrheit der 'da serbo'-Sorten effektiv segregieren und die Populationsstruktur im Zusammenhang mit sowohl Sortentyp als auch geografischer Herkunft widerspiegeln. Der Kopplungsungleichgewicht (LD) zeigte einen schnellen Zerfall über einen genomischen Bereich von weniger als 5 kb. Die Untersuchung von SNPs mit Allelen niedriger Häufigkeit (MAF) in 'da serbo'-Materialien offenbarte eine hochfrequente Mutation in Genen, die mit Stressresistenz assoziiert sind, wie CTR1 und JAR1, die an der Fruchtreifung beteiligt sind. Schließlich wurden durch die Nutzung einer Auswahl von 58 Kernmaterialien, die einen signifikanten Teil der genetischen Vielfalt repräsentieren, wesentliche Merkmale weiterentwickelt. Die genetischen Signaturen des 'da serbo'-Genmaterials, das mit Selektion im Mittelmeerraum kultiviert wurde, werden in dieser Arbeit offenbart. Darüber hinaus bietet sie neue Perspektiven auf das langlebige "da serbo"-Genmaterial und etabliert es als eine reiche Quelle für Gene der Stressresistenz.
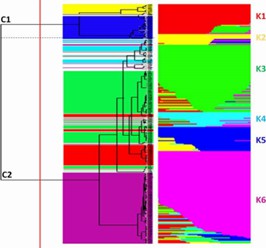 Abb. 1 Schätzung der genetischen Vielfalt in 288 Tomatenzugängen mittels ddRAD.
Abb. 1 Schätzung der genetischen Vielfalt in 288 Tomatenzugängen mittels ddRAD.
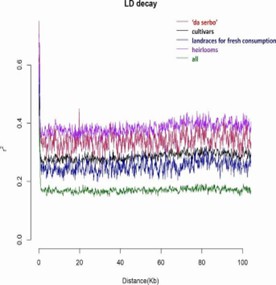 Abb. 2 Abnahme des Linkage-Disequilibrium (LD) und Vergleich.
Abb. 2 Abnahme des Linkage-Disequilibrium (LD) und Vergleich.
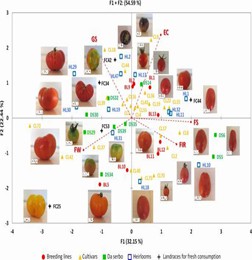 Abb. 3 Ladeplot der ersten (F1) und zweiten (F2) Hauptkomponenten, die die Variation der wichtigsten Fruchteigenschaften in den Zugangsstämmen des Mini-Kernsatzes zeigt, der aus ddRAD SNP-Daten von 288 kultivierten Tomatengenotypen entwickelt wurde.
Abb. 3 Ladeplot der ersten (F1) und zweiten (F2) Hauptkomponenten, die die Variation der wichtigsten Fruchteigenschaften in den Zugangsstämmen des Mini-Kernsatzes zeigt, der aus ddRAD SNP-Daten von 288 kultivierten Tomatengenotypen entwickelt wurde.
Fazit
Durch die Verwendung von ddRAD-seq und dem neuesten Tomatengenom identifizierten die Autoren 2.297 neue Zielgene mit SNPs im mediterranen 'da serbo'-Genpool. Diese Studie zeigt eine geografische Unterscheidung zwischen mediterranen Landrassen und hebt SNPs in neuartigen Genregionen hervor, die mit Stress- und Hormonwegen in Verbindung stehen. Eine Mini-Core-Kollektion vielfältiger Genotypen wurde für die Zucht bereitgestellt und bietet ein wertvolles Genreservoir für zukünftige präzise Züchtung und genomweite Assoziationsstudien.
Referenz
- Esposito, S., Cardi, T., Campanelli, G. et al. ddRAD-Sequenzierung-basierte Genotypisierung zur Analyse der Populationsstruktur in kultivierten Tomaten liefert neue Einblicke in die genomische Vielfalt des mediterranen 'da serbo'-Typs mit langer Haltbarkeit. Gartenbau Forschung 7, 134 (2020).
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Einsatz von Biostimulanzien zur Minderung von Wasserstress in zwei Hartweizen (Triticum durum Desf.) Genotypen mit unterschiedlicher Trockenstress-Toleranz
Journal: Pflanzenstress
Jahr: 2024
Die Restriktions-Modifikationssysteme von Clostridium carboxidivorans P7
Zeitschrift: Mikroorganismen
Jahr: 2023
Im Land der Blinden: Außergewöhnliche subterranne Spezialisierung kryptischer troglobitischer Spinnen der Gattung Tegenaria (Araneae: Agelenidae) in Israel
Zeitschrift: Molekulare Phylogenetik und Evolution
Jahr: 2023
Genetische Modifikatoren des oralen Nikotinkonsums bei Chrna5-Nullmutantenmäusen
Zeitschrift: Front. Psychiatrie
Jahr: 2021
Eine hochdichte genetische Verknüpfungskarte und QTL-Identifizierung für Wachstumsmerkmale bei Dunkel-Kob (Argyrosomus japonicus)
Journal: Aquakultur
Jahr: 2024
Genomische und chemische Beweise für lokale Anpassung an die Resistenz gegenüber verschiedenen Herbivoren in Datura stramonium
Zeitschrift: Evolution
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


