
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben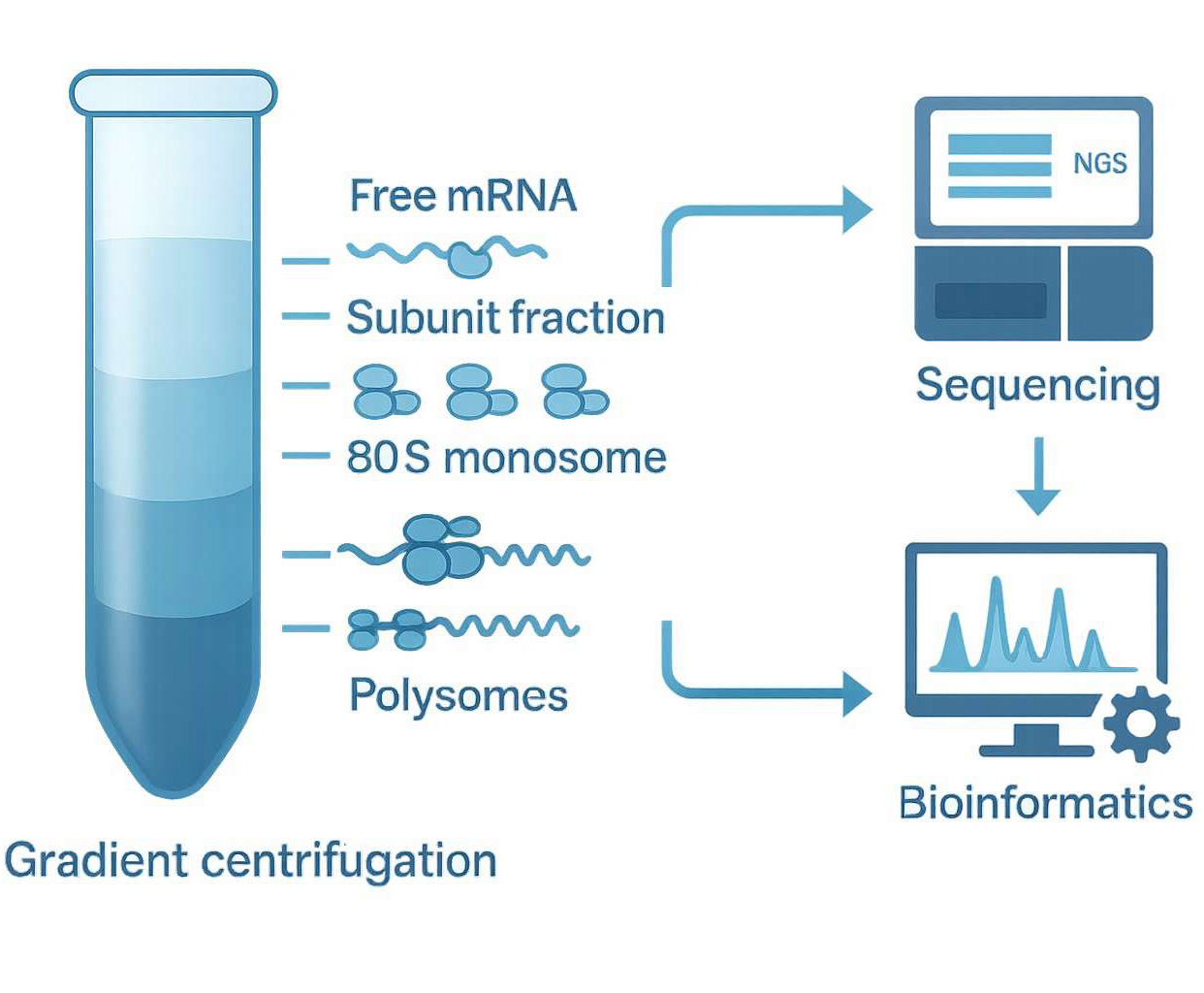
Was ist Polysome-Seq?
In der modernen Molekularbiologie hat sich die Forschung zur Genexpression weit über die bloße Quantifizierung von mRNA-Spiegeln hinaus entwickelt. Der Übersetzungsprozess – bei dem Ribosomen mRNA-Baupläne in Proteine dekodieren – macht über die Hälfte aller genregulatorischen Ereignisse aus und hat einen tiefgreifenden Einfluss auf das Zellverhalten, die Proteinfunktion und die Krankheitsentwicklung. Dennoch reichen traditionelle transkriptomische Methoden oft nicht aus, um die wahre Landschaft der Proteinsynthese offenzulegen, wodurch Lücken zwischen dem gemessenen mRNA-Reichtum und der tatsächlichen Proteinproduktion entstehen.
Polysom-Sequenzierung (Polysome-seq) überbrückt diese kritische Lücke. Durch die Kombination von Polysomen-Profiling mit Hochdurchsatz-Sequenzierung bietet Polysome-seq Forschern eine umfassende, quantitative Momentaufnahme darüber, wie Ribosomen mit Tausenden von mRNAs unter verschiedenen biologischen Bedingungen interagieren. Es enthüllt dynamische Einblicke in die translationale Regulation und ermöglicht eine präzise Untersuchung der Genexpression auf der Ebene, auf der Proteine – die letztendlichen Effektoren der zellulären Funktion – tatsächlich produziert werden.Wie Polysom-Profiling funktioniert
Polysom-Profiling ist eine analytische Methode, die zytoplasmatische RNA basierend auf der Anzahl der an jedes mRNA-Molekül gebundenen Ribosomen trennt. Durch die Verwendung von Sukrosegradienten-Ultrazentrifugation werden zelluläre Lysate in verschiedene Schichten fraktioniert, die Folgendes repräsentieren:
- Freies mRNA (nicht an der Translation beteiligt)
- 40S- und 60S-Ribosomenuntereinheiten
- 80S Monosomen (einzelne Ribosomen)
- Leichte und schwere Polysomen (mehrere Ribosomen, die eine einzelne mRNA übersetzen)

Ein Vergleich von translationalen Omics-Technologien
Die translationale Forschung hat sich erweitert und umfasst mehrere Hochauflösungstechnologien, die jeweils einzigartige Einblicke bieten:
| Technologie | Kernfokus | Wesentliche Vorteile | Einschränkungen |
|---|---|---|---|
| Polysom-Profilierung / Polysom-seq | Misst die Ribosomenbesetzung, um die Übersetzungseffizienz abzuleiten. | - Effizienzmessung der direkten Übersetzung - Beibehaltung längerer RNA-Fragmente für nachfolgende Analysen |
- Größere Stichprobe erforderlich - Keine ribosomalen Positionsdaten |
| Ribo-seq (Ribosomen-Profiling) | Karten präzise Ribosomenpositionen auf mRNA mit Kodonauflösung. | - Erkennt Startstellen, ORFs, uORFs - Enthüllt translativen Stillstand und Dynamik |
- Technisch komplex - Kann aktive von gestoppten Ribosomen nicht unterscheiden. |
| RNC-seq (Ribosomen-Nascent-Chain-Komplex-Sequenzierung) | Erfasst vollständige mRNAs, die an Ribosomen gebunden sind. | - Bewahrt die gesamte mRNA-Struktur - Erkennt alternative Splicing-Isoformen |
- Fehlt an ribosomaler Positionsinformation - Niedrigere Auflösung der Übersetzungsdynamik |
| Disome-seq | Erkennt Ribosomenkollisionen und translationsbedingte Pausen. | - Beleuchtet co-translationalen regulatorischen Ereignisse | - Spezialisierte, neuere Technik mit weniger Anwendungen |
| TRAP-seq | Isoliert ribosomengebundene mRNAs in spezifischen Zelltypen über markierte Ribosomen. | - Zell- oder gewebespezifische Translationsprofilierung | - Benötigt transgene Modelle - Mögliche Beeinträchtigung der Ribosomenfunktion |
Unter diesen Technologien, Polysom-Sequenzierung hebt sich als idealer Kompromiss hervor – es bewahrt die RNA-Integrität, zeigt die Übersetzungseffizienz im gesamten Transkriptom und ermöglicht eine integrative Analyse zusammen mit Transkriptomik, Epitranskriptomik und Proteomik.
Wir bieten ein umfassendes Angebot an translationalen Profilierungsdiensten an, einschließlich Polysome-seq, Ribo-seq, RNC-seq, Disome-seq und TRAP-seq, um den unterschiedlichen Forschungsbedürfnissen in allen Bereichen der Molekularbiologie gerecht zu werden..
Unser Polysom-Sequenzierungs-Workflow: Von der Probe zur Erkenntnis
1. Beratung & Experimentelles Design
Jedes Projekt beginnt mit einer detaillierten Diskussion zwischen unserem wissenschaftlichen Team und Ihrer Forschungsgruppe, um:
- Definieren Sie Ihre biologischen Fragen und Hypothesen.
- Wählen Sie geeignete experimentelle Bedingungen, Kontrollen und Wiederholungen aus.
- Wählen Sie zwischen Einzel-Fraktion oder Multi-Fraktion Polysom-Sequenzierungsstrategien.
- Richten Sie Ihr Projekt an Publikationsstandards aus.
2. Probenvorbereitung & Ribosomenstabilisierung
- Zellen oder Gewebe werden entnommen und mit Inhibitoren der Translationserweiterung (z.B. Cycloheximid) behandelt, um Ribosomen an Ort und Stelle einzufrieren.
- Schnelle Verarbeitung unter RNase-freien Bedingungen bewahrt Ribosomen-mRNA-Komplexe und verhindert deren Abbau.
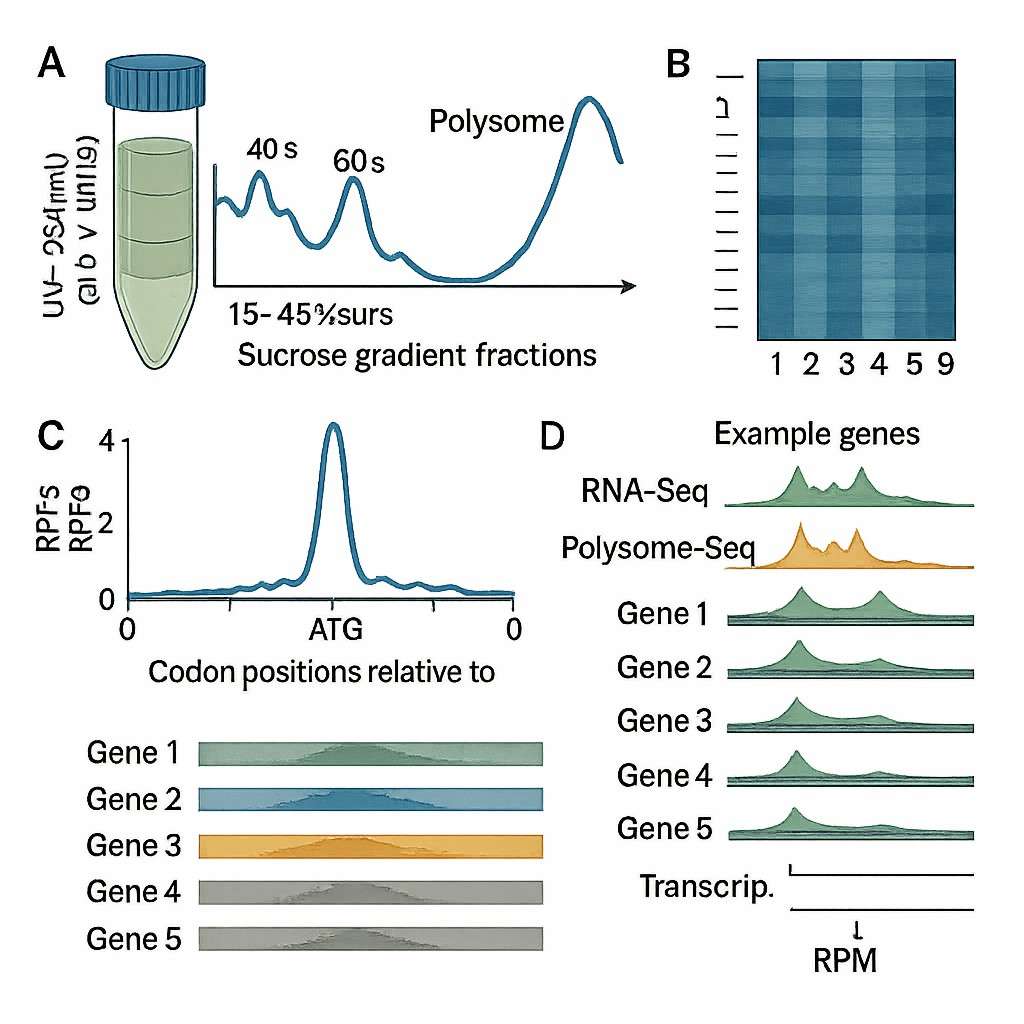
3. Polysom-Profilierung mittels Sukrosegradienten-Ultrazentrifugation
- Zytoplasmatische Extrakte werden auf einen linearen Saccharosegradienten (typischerweise 10-50%) geschichtet.
- Ultrazentrifugation trennt mRNA-Ribosomen-Komplexe basierend auf der Dichte und liefert Fraktionen, die unterscheiden.
- Die UV-Absorptionsprofilierung definiert präzise die Peaks, die jeder Fraktion entsprechen.
4. Fraktionensammlung & RNA-Extraktion
Einzelne Gradientfraktionen werden gesammelt, entweder:
- Als eine einzelne "gebündelte Polysomfraktion" (für kosteneffizientes globales Profiling).
- Oder als mehrere Fraktionen (leichte vs. schwere Polysomen) für eine tiefere Auflösung von translationalen Verschiebungen.
- RNA wird aus jeder Fraktion extrahiert, um eine hohe Integrität für die nachgelagerte Sequenzierung sicherzustellen.
5. Bibliotheksvorbereitung & Next-Generation-Sequenzierung
RNA unterliegt:
- rRNA-Depletion oder Poly-A-Selektion (je nach Studienziel).
- Fragmentierung und Adapterligierung.
- Reverse Transkription und Bibliotheksamplifikation.
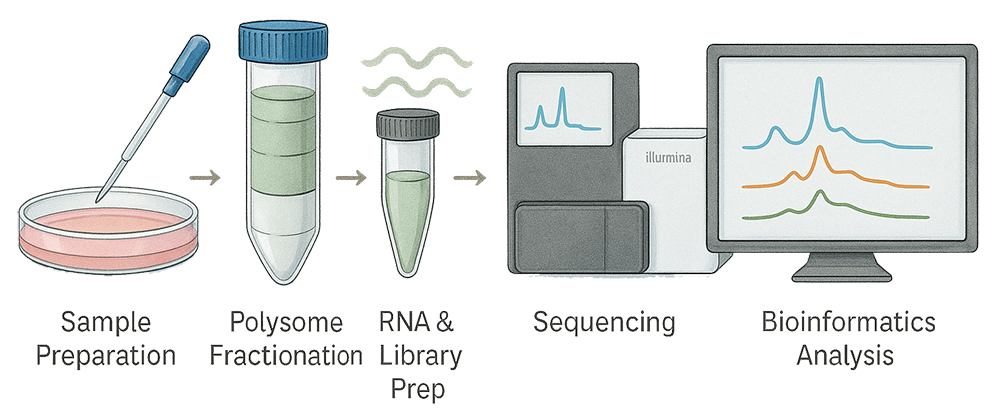
Bioinformatische Analyse und Dateninterpretation
Exzellenz in der Bioinformatik
- Strenge Qualitätskontrolle und Filterung
- Ausrichtung an Referenzgenomen oder Transkriptomen
- Transkriptquantifizierung über Fraktionen hinweg
- Berechnung der Übersetzungseffizienz (TE)
- Differenzielle Übersetzungsanalyse zwischen Bedingungen
- Integration mit:
- RNA-Seq
- Epitranskriptomik (z. B. m6A, m7G)
- Proteomik
Liefergegenstände umfassen:
- Veröffentlichungsfertige Diagramme und Abbildungen
- Umfassende statistische Berichte
- Fachkundige Interpretation, die auf Ihre Studienziele zugeschnitten ist
Musteranforderungen
| Probenart | Mindestbetrag | Notizen |
|---|---|---|
| Mammalianzelllinien | ≥ 4 × 10⁷ Zellen | Kultiviert unter Standardbedingungen; Überkonfluenz vermeiden. |
| Tiergewebe | ≥ 400 mg | Sofort nach der Sektion schockgefrieren. |
| Pflanzengewebe | ≥ 400 mg | Entfernen Sie überschüssiges Wasser, bevor Sie es einfrieren. |
| Bakterienkulturen | ≥ 4 × 10⁷ Zellen | Ernten Sie während der Log-Phase für optimale Ribosomenaktivität. |
| Pilzkulturen | ≥ 4 × 10⁷ Zellen | Stellen Sie eine ordnungsgemäße Homogenisierung vor der Lyse sicher. |
| Isolierte Ribosomenkomplexe | Variable; anfragen | Benutzerdefinierte Protokolle verfügbar für spezialisierte Projekte. |
Probenhandhabungsrichtlinien
- Sofort nach der Ernte Proben in flüssigem Stickstoff schockgefrieren.
- Bei -80°C lagern bis zum Versand.
- Versenden Sie auf Trockeneis, um die RNA-Integrität zu erhalten.
- Kennzeichnen Sie alle Röhrchen deutlich und stellen Sie ein detailliertes Probenblatt zur Verfügung.
- Vermeiden Sie wiederholte Frost-Tau-Zyklen.
Was Sie von unserem Polysomen-Sequenzierungsdienst erhalten werden
- Roh-FASTQ-Dateien aus der Hochdurchsatz-Sequenzierung.
- BAM-Alignmentsdateien, die auf Referenzgenome oder Transkriptome abgebildet sind.
- Quantifizierte Genexpressionsdaten über Polysomfraktionen.
- Übersetzungseffizienz (TE) Berechnungen für jedes Transkript.
- Listen von Genen, die eine unterschiedliche Translation zwischen Bedingungen zeigen.
- Polysomprofil-Diagramme, die die Ribosomenbelegung veranschaulichen.
- Vulkanplots und Heatmaps zur Visualisierung von Translationsänderungen.
- Optionale Multi-Omics-Integration mit RNA-Seq, Epitranskriptomik oder Proteomik.
- Ein umfassender technischer Bericht mit Methoden, Ergebnissen und Interpretationen.
- Direkter Zugang zu unseren Wissenschaftlern für Datenüberprüfungen und Projektdiskussionen.
Warum sollten Sie unseren Polysom-Sequenzierungsdienst wählen?
Unübertroffene Übersetzungsinsights
- Gehen Sie über die mRNA-Häufigkeit hinaus, um zu sehen, wie Gene tatsächlich auf der Ebene der Proteinsynthese exprimiert werden.
- Erfassen Sie die globalen Übersetzungsdynamiken und identifizieren Sie regulatorische Engpässe bei Krankheiten, Stressreaktionen und Entwicklungsprozessen.
Flexibilität für vielfältige Forschungsziele
- Einzel- oder Mehrfachfraktionsoptionen für eine maßgeschneiderte Analysevertiefung.
- Kompatibel mit einer Vielzahl von Organismen, von Säugetierzellen über Pflanzen bis hin zu Mikroben.
- Ultra-niedrig-input Protokolle verfügbar für wertvolle oder begrenzte Proben.
Nahtlose Multi-Omics-Integration
Integrieren Sie Polysomdaten mit:
- Transkriptomik (RNA-Seq)
- Epitranskriptomik (z. B. m6A, m7G Modifikationen)
- Proteomik
- Erhalten Sie einen ganzheitlichen Überblick über die Regulation der Genexpression.
Veröffentlichungsbereite Ergebnisse
- Hochwertige Abbildungen und statistische Analysen, die für wissenschaftliche Zeitschriften erstellt wurden.
- Klare Dokumentations- und Methodensektionen, die für die Einbeziehung in ein Manuskript geeignet sind.
Expertenwissenschaftliche Unterstützung
- Direkter Zugang zu Doktoranden mit umfangreicher Erfahrung in der translationale Forschung.
- Personalisierte Anleitung von der Versuchsplanung bis zur Dateninterpretation.
- Schnelle Reaktionszeiten, um Ihr Projekt voranzubringen.
Nachgewiesene Erfolgsbilanz
- Unterstützte Hunderte von Veröffentlichungen in peer-reviewed Zeitschriften.
- Vertraut von Forschern weltweit in der Akademie, Pharma und Biotechnologie.
Anwendungen der Polysomen-Sequenzierung
Krebsforschung – Entdecken Sie die translationale Umprogrammierung, die das Tumorwachstum und die Therapieresistenz vorantreibt.
RNA-Modifikationsstudien – Entdecken Sie, wie m6A, m7G und andere Modifikationen die Übersetzungseffizienz beeinflussen.
Neurobiologie – Untersuchen Sie die translationale Kontrolle in der neuronalen Entwicklung, Plastizität und neurodegenerativen Erkrankungen.
Pflanzenwissenschaft – Untersuchen Sie Stressreaktionen, Entwicklung und Ertragseigenschaften auf translationaler Ebene in Pflanzen.
Metabolische Forschung – Untersuchen Sie Übersetzungsverschiebungen bei Stoffwechselstörungen und Nährstoffwahrnehmung.
Stammzell-Differenzierung – Erforschen Sie translativer Landschaften, die Zellschicksalsentscheidungen leiten.
Mikrobielle Physiologie – Analysiere die translationale Regulation von Bakterien und Pilzen unter Umweltveränderungen.
Nicht-kodierende RNA Übersetzung – Verborgene Peptide aus lncRNAs, circRNAs und anderen ncRNAs erkennen.
Stressreaktionsmechanismen – Profil globale Translationsänderungen unter Hitzeschock, oxidativem Stress oder Hypoxie.
Studien zu Wirkmechanismen von Medikamenten – Bewerten Sie, wie therapeutische Verbindungen die Translationseffizienz und das Ribosomenengagement beeinflussen.
Demonstrationsergebnisse
Polysome Seq FAQs
1. Was ist Polysome-Seq und wie funktioniert es?
Polysome-Seq kombiniert Polysom-Profiling mit RNA-Sequenzierung, um den translationalen Status im gesamten Transkriptom zu bewerten – indem leichte und schwere ribosomale Fraktionen getrennt und deren mRNA-Gehalt zur Analyse der Translationseffizienz quantifiziert wird.
2. Wie unterscheidet sich Polysome-Seq von Ribo-Seq?
Polysome-Seq profiliert die Anzahl der Ribosomen auf jeder mRNA und bietet einen Überblick über die globale Translation. Im Gegensatz dazu kartiert Ribo-seq die Positionen der Ribosomenfußabdrücke mit Codonauflösung – ideal zum Nachweis von Startstellen, uORFs und pausierter Translation.
3. Kann Polysome-Seq die Translation von nicht-kodierenden RNAs nachweisen?
Ja. Durch die Sequenzierung längerer mRNA-Fragmente aus Polysomfraktionen kann Polysome-Seq aktiv übersetzte lncRNAs, circRNAs und andere ncRNAs erfassen und verborgene Peptide aufdecken.
4. Welche Probenarten sind mit Polysome-Seq kompatibel?
Zu den häufigen Proben gehören kultivierte Zellen, tierische oder pflanzliche Gewebe, Bakterien, Pilze und sogar gereinigte Ribosomenkomplexe. Eine ordnungsgemäße Probenhandhabung und Stabilisierung der Ribosomen sind entscheidend.
5. Wird spezielles Equipment benötigt?
Ja. Die Polysom-Profilierung basiert auf Ultrazentrifugation, Gradientfraktionierung und UV-Detektion. Diese Schritte erfordern technisches Fachwissen und hochwertige Reagenzien – genau das, was unser Team bietet.
6. Welche bioinformatischen Analysen sind enthalten?
Unsere Pipeline umfasst Daten-QC, Genome-/Transkriptom-Ausrichtung, Transkriptquantifizierung, TE-Berechnung, differenzielle Übersetzungsanalyse und Visualisierungen. Eine Integration mit RNA-Seq, Epitranskriptomik oder Proteomik ist ebenfalls verfügbar, um ein umfassendes translationales Profil zu erstellen.
7. Was sind die Einschränkungen von Polysome-Seq?
Potenzielle Herausforderungen umfassen hohe Anforderungen an die Stichprobengröße und eine moderate RNA-Rückgewinnungseffizienz. Darüber hinaus werden keine positionsspezifischen Ribosomdaten bereitgestellt – im Gegensatz zu Ribo-seq.
8. Können Polysome-Seq und Ribo-Seq zusammen verwendet werden?
Absolut. Die Kombination beider Methoden ergibt eine ganzheitliche Sicht: Polysome-Seq zeigt die translationale Aktivität, während Ribo-Seq Details zur Ribosomenpositionierung und nicht-kanonischen Translationsevents bietet.
9. Wie werden Polysomenprofil-Daten interpretiert?
Profile zeigen die Ribosomenverteilung über mRNA anhand von UV-Absorptionsspitzen. Das Verhältnis von Polysomen zu Monosomen weist auf die translativen Aktivitäten hin. Die anschließende Sequenzierung ermöglicht eine quantitative Analyse der Translationseffizienz.
10. Welche Qualitätskontrollmaßnahmen sind vorhanden?
Wir führen strenge Kontrollen in jeder Phase durch – Reproduzierbarkeit des UV-Profils, RNA-Integrität (RIN-Score), Qualitätsmetriken der Bibliothek und bioinformatische Qualitätskontrolle. Diese gewährleisten zuverlässige, hochwirksame Ergebnisse.
Polysom-Seq Fallstudien
Titel: METTL5 stabilisiert c-Myc, indem es die Translation von USP5 erleichtert, um den Glukosestoffwechsel umzuprogrammierten und das Fortschreiten des hepatozellulären Karzinoms zu fördern.
Bitte geben Sie den Text an, den Sie übersetzen möchten. Xia et al., Krebs-Kommunikation, 2023
Impact-Faktor: 20,1
Studienhintergrund
- METTL5 ist eine 18S rRNA Methyltransferase.
- Wissenschaftler vermuteten, dass METTL5 Krebs fördern könnte, indem es beeinflusst, wie bestimmte mRNAs in Proteine übersetzt werden.
- Der Fokus lag auf c-Myc, einem kritischen Onkogen, und USP5, einer Deubiquitinase, die c-Myc stabilisiert.
Wichtige Forschungsfragen
- Beeinflusst METTL5 die c-Myc-Proteinspiegel durch translationaler Regulation?
- Wie beeinflusst dies den Stoffwechsel von Krebszellen?
Verwendete Methoden
✅ Polysom-Profilierung + RNA-Seq
- Getrennte mRNAs in Fraktionen basierend auf der Ribosomenbeladung.
- Sequenzierte polysomgebundene mRNAs zur Bewertung der Translationseffizienz.
✅ qRT-PCR und Western Blot
- Bestätigte Änderungen in RNA-Spiegeln und Proteinspiegeln.
✅ ChIP-Analyse und Metabolomik
- Untersuchte regulatorische Mechanismen und metabolische Veränderungen.
✅ Patient-abgeleitete Xenografts (PDX)
- Validierte Ergebnisse in Tier-Tumormodellen.
Wesentliche Ergebnisse
- METTL5 reguliert die Translation von USP5-mRNA nach oben, was zu höheren USP5-Proteinspiegeln führt.
- USP5 stabilisiert das c-Myc-Protein, indem es dessen Abbau verringert.
- Erhöhtes c-Myc fördert die gesteigerte Expression von glykolytischen Genen (z.B. LDHA, ENO1).
- Der Knockdown von METTL5 führt zu einem reduzierten Tumorwachstum bei Mäusen.
Auswirkungen der Polysom-Sequenzierung
Die Polysomen-Sequenzierung war entscheidend, weil:
- Standard-RNA-Seq allein würde die erhöhte Übersetzungseffizienz von USP5 nicht offenbaren.
- Nur durch die Analyse ribosomenbeladener Fraktionen konnten die Forscher feststellen, wie METTL5 die Produktion von USP5 steigert.
- Helfen Sie dabei, RNA-Methylierung (über METTL5) mit metabolischer Umprogrammierung zu verknüpfen – einem Schlüsselmerkmal von Krebs.
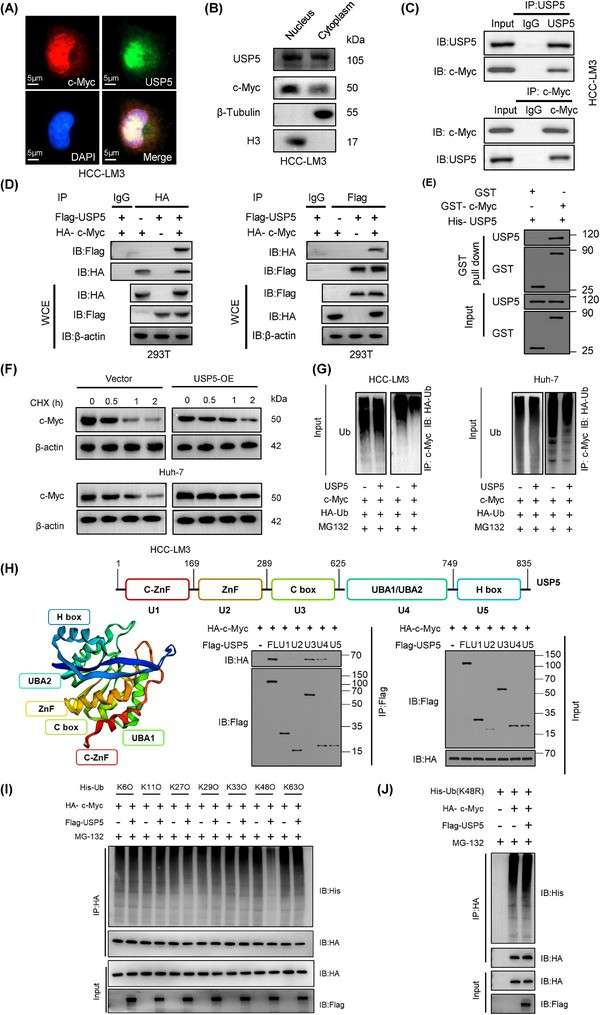 USP5 ist das Deubiquitinisierungsenzym für c-Myc.
USP5 ist das Deubiquitinisierungsenzym für c-Myc.
Warum es wichtig ist
✅ Polysome-seq deckt translativen Regulation auf, die allein durch Transkriptomik unsichtbar bleibt.
✅ Ermöglicht die Entdeckung therapeutischer Ziele wie METTL5.
✅ Zeigt echte Auswirkungen in Krankheitskontexten, von molekularen Mechanismen bis zu Tumorergebnissen.


