
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Vollständige Phagen- und Plasmid-Sequenzierungsdienste
CD Genomics bietet umfassende Plasmid- und Phagen-Sequenzierungsdienste an. Unsere fortschrittlichen bioinformatischen Pipelines unterstützen die de novo Assemblierung, wodurch die Notwendigkeit von Referenzgenomen entfällt und eine gründliche und präzise Analyse sowohl von Plasmiden als auch von Bakteriophagen ermöglicht wird.
Was ist die vollständige Phagen-/Plasmid-Sequenzierung?
Plasmide sind zirkuläre DNA-Moleküle, die unabhängig in bakteriellen Zellen existieren und oft Gene tragen, die ihren Wirten vorteilhafte Eigenschaften wie Antibiotikaresistenz oder verbesserte Stoffwechselfunktionen verleihen. Diese Plasmide sind entscheidend für den horizontalen Gentransfer, der es Bakterien ermöglicht, sich schnell an Umweltbedingungen und Veränderungen anzupassen. Ebenso sind Phagen, oder Bakteriophagen, Viren, die gezielt Bakterien angreifen und eine entscheidende Rolle beim bakteriellen Gentransfer und der Populationsdynamik spielen. Ihre Wechselwirkungen mit bakteriellen Wirten beeinflussen die mikrobielle Vielfalt und das ökologische Gleichgewicht.
Ein umfassendes Verständnis der genetischen Struktur von Plasmiden und Phagen ist entscheidend, um ihre Rollen in mikrobiellen Ökosystemen und ihren Einfluss auf Eigenschaften wie Antibiotikaresistenz zu begreifen. Die vollständige Sequenzierung von Plasmiden/Phagen umfasst das Entschlüsseln der gesamten genetischen Sequenz von Plasmiden und Phagen und bietet einen umfassenden Überblick über ihre genetische Landschaft. Diese Methode ermöglicht es Forschern, sowohl die essentiellen Gene als auch die accessory Elemente zu identifizieren, die zur Anpassungsfähigkeit und pathogenen Potenzial des Organismus beitragen. Durch diese detaillierte genetische Kartierung gewinnen wir tiefere Einblicke in die mikrobielle Evolution und die Prozesse des Gentransfers, die entscheidend für den Fortschritt der Forschung in der Biotechnologie und Umweltwissenschaft sind.
Unsere gesamten Phagen-/Plasmid-Sequenzierungsmethoden
Plasmide und Phagen weisen eine beträchtliche Variabilität in Größe und Komplexität auf, wobei Plasmide oft einen hohen GC-Gehalt, komplexe Strukturen oder umfangreiche repetitive Regionen zeigen, während Phagen ebenfalls unterschiedliche genetische Architekturen und variable Genomgrößen aufweisen können. Diese Komplexitäten haben traditionelle Sanger-Sequenzierungsmethoden historisch sowohl teuer als auch anfällig für hohe Ausfallraten gemacht, verbunden mit relativ langsamen Bearbeitungszeiten.
| Aspekt | Sanger-Sequenzierung | Gesamte Plasmid-/Phagen-SequenzierungNGS) |
|---|---|---|
| Methodik | Kettenabbruchmethode | Nutzen von Next-Generation Sequencing (NGS) oder Long Read Sequencing-Technologien |
| Sequenzlänge | Geeignet für kürzere Fragmente (bis zu 1.000 bp) | In der Lage, gesamte Plasmide und Phagen unabhängig von Größe oder Komplexität zu sequenzieren. |
| Geschwindigkeit | Die Sanger-Sequenzierung kann längere Bearbeitungszeiten erfordern und eignet sich besonders für kleinere Projekte. | Die vollständige Plasmid-/Phagen-Sequenzierung mit NGS-Technologien bietet eine schnellere Bearbeitungszeit, die sich ideal für Hochdurchsatzanwendungen eignet. |
| Kosten | Kann für größere Plasmide aufgrund mehrerer Sequenzierungsreaktionen kostenintensiv sein. | Im Allgemeinen kosteneffektiver, insbesondere für größere Plasmide und Phagen, da es weniger Sequenzierungsreaktionen erfordert. |
| Genauigkeit | Die Sanger-Sequenzierung ist bekannt für ihre hohe Präzision und minimalen Fehlerquoten. | Die vollständige Plasmid-/Phagen-Sequenzierung weist eine außergewöhnliche Genauigkeit auf und integriert fortschrittliche Fehlerkorrekturmethoden. |
| Anwendbarkeit | Die Sanger-Sequenzierung ist effektiv für kleinere Plasmide, kann jedoch bei größeren, komplexen oder repetitiven Strukturen auf Herausforderungen stoßen. | Die vollständige Plasmid-/Phagen-Sequenzierung ist äußerst vielseitig und kann eine breite Palette von Plasmid- und Phagengrößen sowie -komplexitäten aufnehmen, was sie für umfassende genomische Analysen geeignet macht. |
CD Genomics setzt fortschrittliche Sequenzierungstechnologien ein, um vollständige Plasmid- und Phagen-DNA-Sequenzen bereitzustellen. Unsere Methoden integrieren sowohl Next-Generation-Sequencing (NGS) als auch Long-Read-Sequenzierungstechnologien, um die unterschiedlichen Strukturen und Größen von Plasmiden und Phagen zu berücksichtigen. Wir nutzen modernste Plattformen wie Illumina für Hochdurchsatz-Sequenzierung und Nanopore oder PacBio für Long-Read-Sequenzierung. Unser Ansatz umfasst eine umfassende Bibliotheksvorbereitung und bioinformatische Analyse, um eine genaue Zusammenstellung und Annotation der gesamten genetischen Sequenz sicherzustellen.
Phagen-Ganzgenom-Sequenzierung
CD Genomics konzentriert sich nun auf das Angebot von Sequenzierungen von Bakteriophagen, wobei sowohl Kurzlese- (Illumina HiSeq) als auch Langlese-Plattformen (PacBio SMRT, Oxford Nanopore) verwendet werden, um zu unterstützen. von neuem und referenzgeführte Assemblierung. Unser optimierter Workflow verarbeitet GC-reiche, wiederholungsdichte oder strukturell komplexe Phagen-Genome und erzeugt hochkontinuierliche Assemblierungen mit funktioneller Genannotation und Einblicken in die Wirt-Interaktionen.
Die Ganzgenomsequenzierung von Phagen unterstützt die Forschung zur Entdeckung von Phagenkandidaten gegen antibiotikaresistente Bakterien, Studien zur phagenbedingten Diversität in der Umwelt und in Lebensmitteln sowie zur evolutionären Analyse von Phagenpopulationen in verschiedenen Ökosystemen.
Vorteile der vollständigen Phagen-/Plasmid-Sequenzierung
Umfassende DeckungBietet eine vollständige Sequenzierung von Plasmiden und Phagen, einschließlich Regionen mit hohem GC-Gehalt und komplexen Strukturen, die traditionelle Methoden möglicherweise übersehen, und gewährleistet einen umfassenden genetischen Überblick.
Hohe GenauigkeitLiefert Sequenzen mit einer Genauigkeit von über 99,9 %, wodurch sichergestellt wird, dass die genetischen Informationen präzise und zuverlässig für weitere Forschungen sind.
KosteneffektivBietet eine kostengünstigere Lösung, insbesondere für große Plasmide und Phagen, mit deutlich niedrigeren Kosten im Vergleich zu traditionellen Sequenzierungsmethoden, was es zu einer kosteneffizienten Wahl für umfangreiche Projekte macht.
Schnelle BearbeitungStellt eine schnelle Verarbeitung und Datenerfassung sicher, beschleunigt die Forschung und ermöglicht zeitnahe Erkenntnisse.
Keine Referenz erforderlichFührt Sequenzierungen ohne die Notwendigkeit vorhandener Referenzgenome durch, ideal für die Erforschung neuartiger oder uncharakterisierter Plasmide und Phagen.
Hohe DurchsatzleistungIn der Lage, mehrere Proben gleichzeitig zu bearbeiten, was die Effizienz steigert und großangelegte Forschungsanstrengungen unterstützt.
Anwendungen der vollständigen Phagen-/Plasmid-Sequenzierung
- Mikrobielle ÖkologieUntersuchen Sie die Rolle von Plasmiden und Phagen innerhalb mikrobieller Gemeinschaften, ihre Interaktionen und ihren Einfluss auf die mikrobielle Vielfalt und die Funktionen von Ökosystemen.
- GenübertragungsstudienUntersuchen Sie die Mechanismen des horizontalen Gentransfers, die durch Plasmide und Phagen vermittelt werden, einschließlich der Verbreitung von Resistenzgenen und anderen funktionalen Eigenschaften.
- Synthetische BiologieNutzen Sie umfassende Plasmidsequenzen, um genetische Konstrukte zu entwerfen und zu verfeinern, und fördern Sie Anwendungen in der synthetischen Biologie.
- PhagenforschungAnalysiere Phagen-Genome, um Strategien für gezielte Interventionen gegen spezifische Bakterienarten zu entwickeln.
- UmweltforschungUntersuchen Sie das Vorkommen und die Funktionen von Plasmiden und Phagen in verschiedenen Umgebungen, wie z.B. Boden, Wasser und verschiedenen mikrobiellen Lebensräumen.
Vollständiger Plasmid-/Phagen-Sequenzierungs-Workflow

Dienstspezifikationen
| Beispielanforderungen Vollständige Plasmid-DNA-Sequenzierung:
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse Wir bieten mehrere maßgeschneiderte bioinformatische Analysen an:
|
Analyse-Pipeline
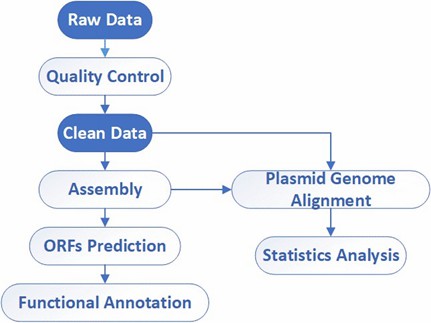
Liefergegenstände
- FASTQ-Dateien
- Leselängen- und Qualitätsbericht
- Versammlungsergebnisse
- Genomische Kreisdiagramm
- Analyse potenzieller Mutationen
- Annotierungsdateien
CD Genomics nutzt die fortschrittlichen Möglichkeiten der Illumina-Plattform für präzises Plasmid-Sequencing, um eine zuverlässige Verifizierung und umfassende Analyse von Plasmid-DNA zu gewährleisten. Darüber hinaus haben wir eine kosteneffiziente, hochdurchsatzfähige Methode mit Nanopore-Technologie entwickelt, um größere Plasmide effizient in ihrer Gesamtheit zu sequenzieren. Indem wir unser Fachwissen auf das Phagen-Sequencing ausdehnen, nutzen wir diese modernen Technologien, um umfassende und genaue genomische Einblicke in Bakteriophagen zu bieten. Mit unserer umfangreichen Erfahrung und modernsten Plattformen sind wir bestrebt, außergewöhnliche Sequenzierungsdienste und hochwertige Ergebnisse zu liefern, die auf die vielfältigen Bedürfnisse unserer Kunden zugeschnitten sind.
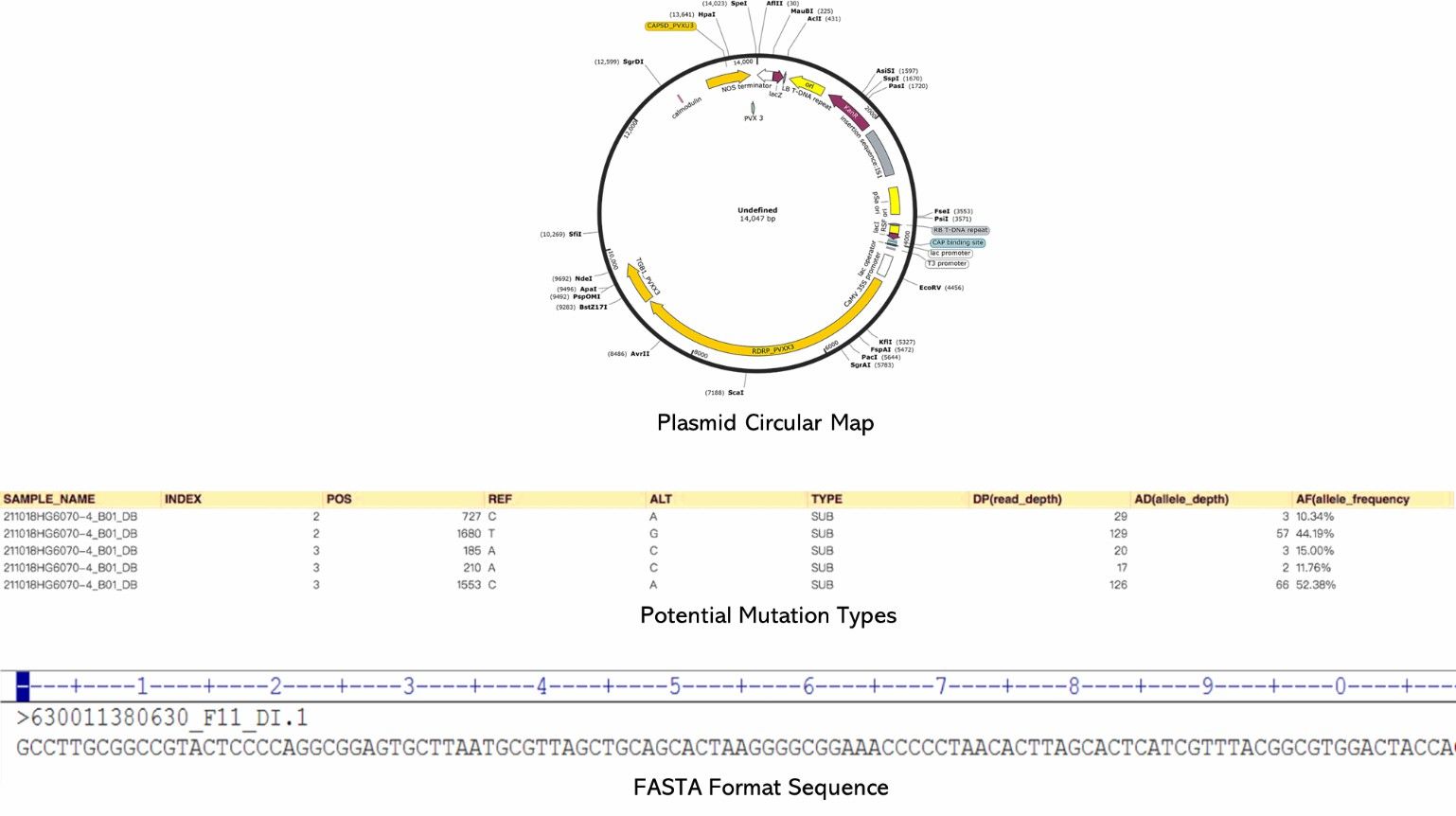
Demo-Ergebnisse
Vollständige FAQs zu Phagen- und Plasmid-Sequenzen
1. Warum ist die vollständige Sequenzierung von Plasmid-DNA notwendig?
- Validierung der Sequenzintegrität: Plasmide können während des Klonens oder der Amplifikation genetischen Veränderungen unterliegen, was potenzielle Fehler oder Mutationen einführt.
- Qualitätskontrolle für Klonierung und Ingenieurwesen: Die Sequenzierung von Plasmiden gewährleistet eine genaue Überprüfung der klonierten Einsätze, das Fehlen von Mutationen und die Integrität des experimentellen Systems.
- Optimierung des experimentellen Designs: Exakte Plasmidsequenzen ermöglichen eine effektive Versuchsplanung.
2. Wie extrahiert man Plasmid-DNA aus Wirtzellen?
Häufige Extraktionsmethoden sind:
- Alkalische Lyse: Zelllyse unter alkalischen Bedingungen zur Freisetzung von Plasmid-DNA.
- Phenol-Chloroform-Extraktion: Verwendung von Phenol und Chloroform zur DNA-Extraktion, um Proteine und Verunreinigungen zu entfernen.
- Säulenbasierte Extraktion: Kommerzielle Säulen zur Reinigung von Plasmid-DNA durch Zentrifugation und Elution.
3. Wie kann die Genauigkeit von Sequenzierungsdaten sichergestellt werden?
Kritische Maßnahmen zur Sicherstellung der Präzision von Sequenzierungsdaten umfassen mehrere wichtige Verfahren:
- Gründliche Probenentnahme: Gewährleistung der Reinheit und Konzentration von Plasmid-DNA.
- Auswahl einer geeigneten Sequenzierungsplattform: Anpassung der Wahl an die experimentellen Anforderungen.
- Implementierung der Datenverarbeitung und Qualitätskontrolle: Einsatz von bioinformatischen Werkzeugen zur Eliminierung von minderwertigen Reads und Sequenzierungsfehlern.
- Validierung durch Sequenzalignment: Abgleich der Ergebnisse mit bekannten Sequenzen zur Untermauerung der Genauigkeit.
4. Was sind die Anwendungen der vollständigen Plasmid-DNA-Sequenzierung?
Die vollständige Sequenzierung von Plasmid-DNA bietet eine Vielzahl von Anwendungen in verschiedenen Bereichen:
- Genklonierung und Expressionsstudien: Erleichterung des Aufbaus effektiver Klonierungsvektoren.
- Forschung zur Antibiotikaresistenz: Entschlüsselung bakterieller Resistenzmechanismen.
- Genfunktionen Studien: Untersuchung spezifischer Genfunktionen innerhalb von Zellen.
- Synthetische Biologie und Biotechnologie: Pionierarbeit bei der Entwicklung von Werkzeugen und Wegen der synthetischen Biologie für die pharmazeutische Produktion, die Umweltrestaurierung und Bioenergieprojekte.
Wenn Sie mehr erfahren möchten, lesen Sie bitte unseren Artikel "Plasmidnachweis und vollständige Plasmid-DNA-Sequenzierung."
5. Welche Faktoren können die Ergebnisse der Plasmid-DNA-Sequenzierung beeinflussen?
Einflussfaktoren umfassen:
- Probenqualität: DNA-Reinheit und Konzentrationsangemessenheit.
- Sequenzierungstechnologie: Angemessenheit der gewählten Plattformen und Technologien.
- Datenverarbeitung: Effektivität der Qualitätskontrolle und der Verarbeitungsverfahren.
- Sequenzkomplexität: Einfluss der Sequenzkomplexität von Plasmiden und repetitiven Elementen auf die Assemblierung.
Vollständige Fallstudien zu Phagen- und Plasmid-Sequenzen
Dynamik der antimikrobiellen Resistenz und genomische Epidemiologie von multiresistenten Erregern Salmonella enterica Serovar Indiana ST17 von 2006 bis 2017 in China
Journal: Msystems
Impactfaktor: 7,324
Veröffentlicht: 21. Juli 2022
Hintergrund
Nontyphoidale Salmonella enterica (NTS) ist ein bedeutender globaler lebensmittelbedingter Krankheitserreger, der zunehmend mit antimikrobieller Resistenz (AMR), insbesondere multiresistenten (MDR) Stämmen, in Verbindung gebracht wird. Diese Resistenz erschwert die Behandlung und stellt eine Herausforderung für die öffentliche Gesundheit dar, was die WHO dazu veranlasst hat, resistente S. enterica für die Entwicklung neuer Antimikrobiaka zu priorisieren. Die genetischen Grundlagen der Resistenz umfassen chromosomale Mutationen und plasmidvermittelte Gene, die zur weit verbreiteten Dominanz bestimmter Stämme beitragen. Neueste Studien in China heben die Prävalenz und die Resistenzmechanismen von MDR S. Indiana hervor und unterstreichen den dringenden Bedarf an effektiven Maßnahmen zur Eindämmung seiner Verbreitung.
Methoden
- 251 SIndiana-Isolate
- Stuhlproben
- Klinische Proben und lebensmittelbezogene Proben
- Antimikrobielle Empfindlichkeitstestung
- DNA-Extraktion
- Whole-Genome-Sequenzierung
- Erkennung von AMR Genotypen
- Analyse von blaCTX-M genomische Standorte
- Phylogenetische Analyse
Ergebnisse
Unter 138 blaCTX-M-positive Isolate, 65,2 % wurden nach Genomstandort klassifiziert. Bemerkenswert, blaCTX-M-14 und blaCTX-M-55 wurden auf Chromosomen identifiziert, während blaCTX-M-15 und blaCTX-M-65 wurden überwiegend von Plasmiden getragen. Menschliche Isolate wiesen eine signifikant höhere Prävalenz von blaCTX-M Positivität (63%) im Vergleich zu lebensmittelbezogenen Isolaten (47%). Die vollständige Sequenzierung von fünf repräsentativen Isolaten enthüllte eine vielfältige Landschaft genetischer Kontexte, die beherbergen blaCTX-M Gene, insbesondere auf IncHI2-Plasmiden. Bemerkenswerterweise wies das Plasmid pIndS104-CTX eine auffällige Ähnlichkeit mit einem chromosomalen Locus in S. Indiana SI43 auf, was auf potenzielle Rekombinationsereignisse zwischen Plasmiden und Chromosomen hindeutet. Diese Studie hebt die Komplexität von hervor. blaCTX-M Verbreitungswege unter Pathogenen und betont die dringende Notwendigkeit weiterer Aufklärung.
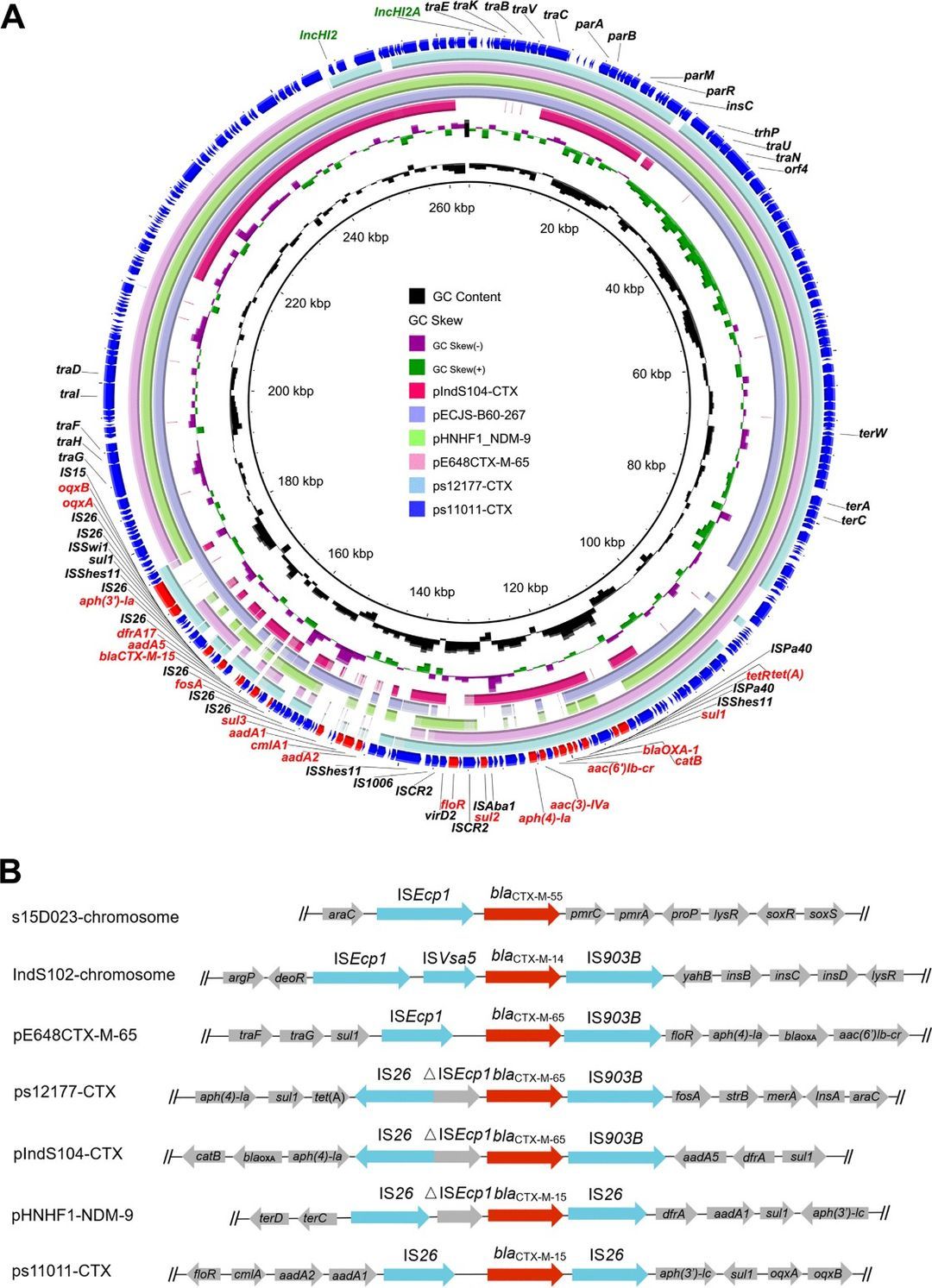 Abb. 1. Zirkulärer Vergleich zwischen blaCTX-M-positive IncHI2-Plasmide in dieser Studie (pIndS104-CTX, ps17177-CTX und ps11011-CTX) und andere ähnliche IncHI2-Plasmide in der NCBI nr-Datenbank.
Abb. 1. Zirkulärer Vergleich zwischen blaCTX-M-positive IncHI2-Plasmide in dieser Studie (pIndS104-CTX, ps17177-CTX und ps11011-CTX) und andere ähnliche IncHI2-Plasmide in der NCBI nr-Datenbank.
Diese landesweite Studie von 251 Isolaten fand eine starke genomische Ähnlichkeit zwischen menschlichen und Hühnisolaten, was darauf hindeutet, dass Hühner eine Quelle für menschliche Infektionen sind. Linie 6 war die resistenteste, wobei IncHI2-Plasmide häufig ESBL- und PMQR-Gene trugen.
Fazit
Diese Studie bietet einen detaillierten Einblick in die rasante Entwicklung der Mehrfachresistenz (MDR) von S. Indiana in den letzten 15 Jahren in China. Einzigartige Mechanismen der antimikrobiellen Resistenz unterscheiden S. Indiana von anderen Serovaren, wobei verschiedene genetische Prozesse zur Entwicklung der Resistenz beitragen, einschließlich chromosomaler Integrationen, der Evolution mobiler Resistenzelemente und sporadischer Akquisition von Resistenzdeterminanten. Die Präsenz vielfältiger Wirt-Nischen, einschließlich verschiedener tierischer Reservoirs, unterstreicht die Bedeutung eines One-Health-Ansatzes für ein effizientes Monitoring und die Kontrolle der Ausbreitung von Resistenzen. Eine kontinuierliche Überwachung, die auf bakterielle Stämme und mobile genetische Elemente abzielt, ist für effektive Kontrollmaßnahmen unerlässlich.
Referenz:
- Du P, Liu X, Liu Y, et al. Dynamik des antimikrobiellen Widerstands und genomische Epidemiologie von multiresistenten Salmonella enterica Serovar Indiana ST17 von 2006 bis 2017 in China. Msysteme, 2022, 7(4): e00253-22.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Unterscheidliche Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die therapieinduzierte Seneszenz.
Zeitschrift: Zellsterben & Krankheit
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generierung eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genomanalysen und Replikationsstudien des afrikanischen grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


