
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Langzeit-Sequenzierung
CD Genomics bietet modernste Technologien und bioinformatische Analyse-Dienstleistungen für Long-Read-Sequenzierung an, die Folgendes umfassen: PacBio SMRT-Sequenzierung und Nanoporen-SequenzierungWir bieten präzise und kosteneffiziente Sequenzierungslösungen für Menschen, Tiere, Pflanzen und mikrobielle Forschung an.
Was ist Long-Read-Sequenzierung?
Long-Read-Sequenzierung, auch bekannt als Sequenzierung der dritten Generation, ist eine Gruppe von boomenden DNA-Sequenzierung Technologien, die die Notwendigkeit traditioneller Sequenzierungstechniken zur DNA-Spaltung und -Amplifikation beseitigen und die gleichzeitige Erkennung langer DNA-Sequenzen von bis zu 100.000 Basenpaaren ermöglichen. Wissenschaftler haben die Bedeutung struktureller Varianten im menschlichen Genom erkannt. Einige von ihnen sind insertionsbedingte Varianten, die die Lese- länge vieler Sequenzierungstechnologien überschreiten; einige sind repetitive Regionen, die die Sequenzanpassung erschweren; und einige sind GC-reiche Regionen, die oft viele Unannehmlichkeiten bei der Untersuchung struktureller Varianten durch Methoden der Kurzlesesequenzierung mit sich bringen.
In den letzten Jahren sind neuartige Technologien entstanden, die lange Lesesequenzen erkennen können und den Forschern neue Strategien und Werkzeuge für die Genom-Analyse bieten. Unter ihnen sind die repräsentativsten und am weitesten verbreiteten PacBio SMRT-Sequenzierung und Nanoporen-SequenzierungLangzeit-Sequenzierungstechnologien können sehr lange Reads erzeugen, was bei herkömmlichen Methoden nicht möglich ist. Next-Generation-Sequenzierung.
PacBio hat die Sequenzierungsplattform von Einzelmolekül-Echtzeit-Sequenzierung (SMRT), das eine maßgeschneiderte Flusszelle mit vielen Zero-Mode-Wellenleitern verwendet, um einzelne Moleküle in Echtzeit zu sequenzieren (ZMW). Die Polymerase ist am Boden des Wells befestigt und ermöglicht es dem DNA-Strang, durch den ZMW zu gelangen. Mit der SMRT-Sequenzierung ist eine Echtzeit-Bildgebung von fluoreszenzmarkierten Nukleotiden möglich, die zusammen mit spezifischen DNA-Vorlagenmolekülen synthetisiert werden. Wenn sich das Gerüst und die Polymerase trennen, ist die Sequenzierungsreaktion abgeschlossen.
Die Kernelemente von Die Technologie von Nanopore Unterteilen in die Bildung von Transmembrankanälen aus Nanoporen, die ionische Ströme durchlassen, und die Messung der Änderungen im Strom. Wenn Moleküle wie DNA oder RNA durch Nanoporen hindurchtreten, verursachen sie Störungen im Strom. Die Informationen über die Änderungen im Strom können verwendet werden, um das Molekül zu identifizieren. Es sequenziert die gesamten DNA/RNA-Moleküle direkt.
Vorteile der Langzeit-Sequenzierung
- Lange durchschnittliche Leselängen und hohe Konsensgenauigkeit
- Verbesserte Genauigkeit für wiederholte Sequenzen und Kopienzahlvariationen
- Genauere Erkennung einer großen Anzahl von Mutationen
- Optimierung des DNA-Extraktionsprotokolls bei Langzeit-Sequenzierung
- Kompatibel mit sowohl Genom- als auch Transkriptomanalysen
- Schnell und erschwinglich
Anwendung von Langzeit-Sequenzierung
- Genfunktion: Konzentrieren Sie sich auf Proben, die spezifische Funktionen tragen, um die Hauptursachen für unterschiedliche Funktionen zu enthüllen.
- Genstruktur: Alternatives Spleißen, APA, Fusionsgen, SSR, CDS-Vorhersage, TSS/TES-Identifikation usw.
- Vollständige Quantifizierung: Finden Sie umfassende und effektive differentielle Gene und identifizieren Sie funktionale Gene.
- Epigenomik: Direkte vollständige Genom- und Transkriptom-Sequenzierung kann Basismodifikationen erkennen.
Long-Read-Sequenzierungs-Workflow
CD Genomics nutzt mehrere Plattformen, um schnelle und präzise Long-Read-Sequenzierungsdienste und bioinformatische Analysen anzubieten. Unsere hochqualifizierten Experten führen ein Qualitätsmanagement durch und befolgen jeden Schritt, um hochwertige Ergebnisse zu gewährleisten. Der allgemeine Ablauf für die Long-Read-Sequenzierung ist unten skizziert.

Dienstspezifikationen
Musteranforderungen
|
|
 Klicken |
Sequenzierungsstrategie
|
 |
Bioinformatikanalyse
|
Analyse-Pipeline
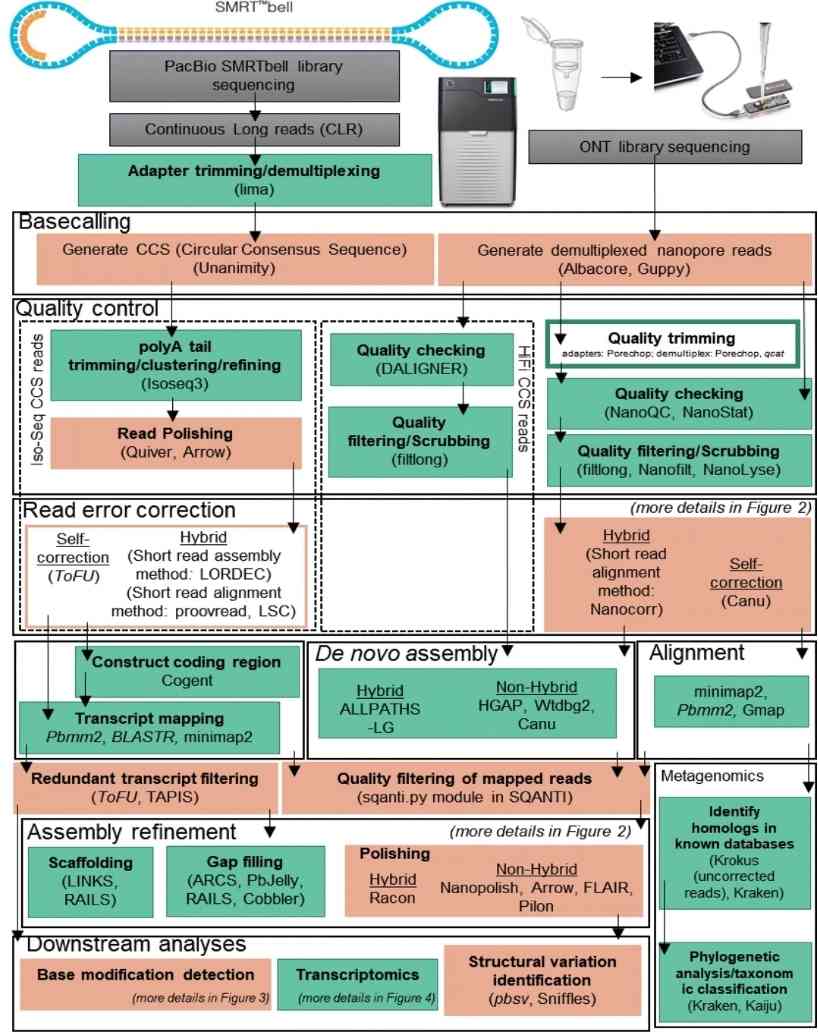 Übersicht über Werkzeuge und Pipelines zur Analyse von Langreads. (Amarasinghe et al., 2020)
Übersicht über Werkzeuge und Pipelines zur Analyse von Langreads. (Amarasinghe et al., 2020)
Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in der Langzeit-Sequenzierung für Ihr Schreiben (Anpassung)
CD Genomics bietet ein umfassendes Servicepaket für Long-Read-Sequenzierung an, das Probenvorbereitung, Bibliothekskonstruktion umfasst, SMRT-Sequenzierung und/oder Nanoporen-Sequenzierungund bioinformatische Analysen. Wir können diese Pipeline auf Ihr Forschungsinteresse zuschneiden. Wenn Sie zusätzliche Anforderungen oder Fragen haben, zögern Sie bitte nicht, uns zu kontaktieren.
Referenz
- Amarasinghe S L, Su S, Dong X, et al. Chancen und Herausforderungen bei der Analyse von Langsequenzierungsdaten. Genombiologie, 2020, 21(1): 30.
Demonstrationsergebnisse
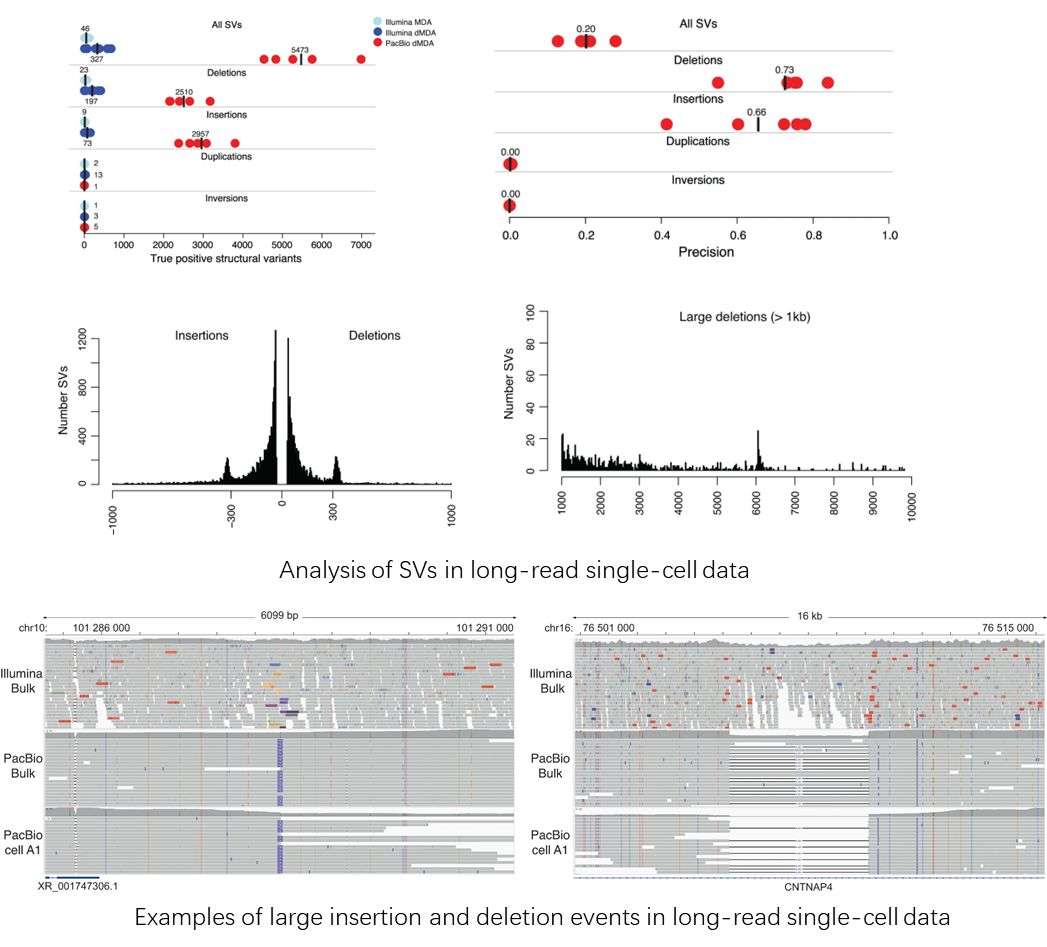 Verwendung von Ergebnissen der Einzelzell-Ganzgenom-Langlesesequenzierung als Beispiel. (Hård et al., 2023)
Verwendung von Ergebnissen der Einzelzell-Ganzgenom-Langlesesequenzierung als Beispiel. (Hård et al., 2023)
Referenz
- Hård J, Mold J E, Eisfeldt J, et al. Langzeit-Whole-Genome-Analyse von menschlichen Einzelzellen[J]. Nature Communications, 2023, 14(1): 5164.
Long-Read-Sequenzierungs-FAQs
1. Was ist der Unterschied zwischen Illumina-Sequenzierung und Langlese-Sequenzierung?
Illumina-Sequenzierung, auch als Kurzlesesequenzierung bezeichnet, liefert kürzere Sequenzlesungen, die typischerweise zwischen 100 und 300 Basenpaaren lang sind. Im Gegensatz dazu bieten Technologien zur Langlesesequenzierung, wie die von Oxford Nanopore Technologien und Pacific Biosciences, erzeugen signifikant längere Reads, die sich von Tausenden bis zu Zehntausenden von Basenpaaren erstrecken. Diese Variabilität in der Read-Länge ermöglicht es der Langzeit-Sequenzierung, die Auflösung komplexer genomischer Regionen, einschließlich repetitiver Sequenzen und struktureller Varianten, zu verbessern, was zu einem umfassenderen genomischen Verständnis im Vergleich zur Illumina-Sequenzierung führt. Die Anwendung der Langzeit-Sequenzierung unterstützt erheblich das Entschlüsseln komplexer bakterieller Genome, insbesondere wenn sie mit kurzen Illumina-Reads durch hybride Assemblierungsmethoden kombiniert wird.
2. Für welche Forschungsbereiche ist das Langzeit-Sequencing geeignet?
Long-Read-Sequenzierung ist in verschiedenen Forschungsbereichen anwendbar innerhalb von Genomikeinschließlich der Humangenetik, Evolutionsbiologie, Mikrobiologie und Botanik. Es ermöglicht Forschern, ein umfassenderes Verständnis der Genomstruktur und -funktion zu erlangen, wodurch Fortschritte in diesen Bereichen vorangetrieben werden.
3. Was sind die Vor- und Nachteile der PacBio-Plattform?
Der PacBio-Plattform verfügt über eine hohe Einzelmolekülgenauigkeit, wobei über 90 % der Sequenzen einen Qualitätswert (Q) von über 30 erreichen, und bietet Sequierungsdaten mit einer Ausrichtungsgenauigkeit, die die von NGS Plattformen. Darüber hinaus bietet es eine durchschnittliche Leselänge, die übersteigt NGS Plattformen um mehr als zwei Größenordnungen. Zu den Nachteilen gehören jedoch deutlich höhere Sequenzierungskosten im Vergleich zu Plattformen wie Novaseq, hohe Gerätekosten und die Unfähigkeit, Sequenzierungsdaten in Echtzeit zu interpretieren, wodurch die Anforderungen an Szenarien, die hohe Aktualität erfordern, nicht erfüllt werden.
4. Was sind die Vor- und Nachteile der ONT-Technologie?
Die Vorteile von ONT-Technologie liegen in seiner hohen Sequenzierungsgeschwindigkeit und den relativ niedrigeren Sequenzierungskosten im Vergleich zu den PacBio-PlattformDarüber hinaus übersteigt die durchschnittliche Leselänge die von NGS Plattformen um mehr als zwei Größenordnungen. Ihre Nachteile ergeben sich jedoch aus systematischen Fehlern bei der Signalverarbeitung, wie Schwierigkeiten bei der Identifizierung von Basen-Homopolymeren, was zu erheblichen Unterschieden sowohl in der Genauigkeit einzelner Moleküle als auch in der Ausrichtungsgenauigkeit im Vergleich zu PacBio- und NGS-Plattformen führt. Diese Einschränkungen begrenzen ihre Anwendungen.
Langzeit-Sequenz-Fallstudien
Die Landschaft der genomischen strukturellen Variation bei indigenen Australiern
Zeitschrift: Natur
Impactfaktor: 45,16
Veröffentlicht: 13. Dezember 2023
Hintergrund
Australien beherbergt Hunderte von Aborigines-Nationen, die jeweils eine reiche kulturelle und sprachliche Vielfalt aufweisen. Trotz umfangreicher Dokumentation ihres kulturellen Erbes ist die genomische Vielfalt der indigenen Australier untererforscht, mit einer erheblichen Unterrepräsentation in globalen genomischen Datenbanken. Das Nationale Zentrum für Indigene Genomik (NCIG) schließt diese Lücke, indem es Aborigines-Gemeinschaften in ethische und gemeinschaftsgeleitete genomische Forschungsprojekte einbezieht. In dieser Studie wurde eine bevölkerungsweite Langlesesequenzierung in Aborigines-Gemeinschaften durchgeführt mit Oxford Nanopore Technologien, die zuvor unentdeckte genomische Variationen und strukturelle Varianten offenbaren, die entscheidend für den Fortschritt der genomischen Medizin sind.
Methoden
Probenvorbereitung:
Speichelprobenentnahme
Frische Blutentnahme
DNA-Extraktion
Sequenzierung:
Bibliothekskonstruktion
Ganzgenom ONT-Sequenzierung
ONT-Datenverarbeitung
Ausrichtung an das Referenzgenom
Erkennung von strukturellen Variationen
Strukturelle Variationswiederholungsklassifizierung
Vergleich zu Anmerkungen
Analyse großer CNVs
Ergebnisse
Das vollständige T2T-chm13-Genomreferenz wurde verwendet, um die Kartierbarkeit und die Erkennung struktureller Varianten (SV) zu bewerten, was die überlegene Ausrichtung und SV-Erkennung von ONT zeigte. Dies ergab 159.912 einzigartige SVs und 136.797 große Indels und identifizierte 11 hochkonfidente Kopienzahlvarianten (CNVs) pro Individuum, was die Vorteile des Langzeit-Sequenzierens für eine detaillierte genomische Profilierung hervorhebt.
 Abb. 1 Langzeit-Sequenzierung in indigenen australischen Gemeinschaften.
Abb. 1 Langzeit-Sequenzierung in indigenen australischen Gemeinschaften.
Um genomische strukturelle Variationen zu charakterisieren, kategorisierten die Autoren Varianten nach Typ, Größe und Kontext. Die meisten Varianten (84,9%) waren repetitive Sequenzen, einschließlich STRs, TRs und mobilen Elementeinsertionen/-deletionen. CNVs waren seltener, deckten jedoch bedeutende Genomregionen ab. Die Varianten waren ungleichmäßig verteilt, mit höherer Dichte in der Nähe von Telomeren, insbesondere bei TR-assoziierten SVs. Die Größe variierte je nach Typ, wobei TR-SVs größer waren als STRs und nicht-repetitive SVs. Mobile Element-SVs zeigten Größenpeaks, die mit wichtigen Wiederholungsfamilien wie Alu, L1 und SVA übereinstimmten. Viele SVs waren einzigartig für diese Studie, insbesondere angesichts der Einbeziehung unterrepräsentierter australischer Gemeinschaften und der Verwendung von Long-Read-Sequenzierung. Im Vergleich zu globalen Datenbanken fanden die Autoren eine Ähnlichkeit von etwa 38% mit annotierten SVs, was auf einen hohen Anteil neuartiger SVs hinweist.
 Abb. 2 Landschaft der genomischen strukturellen Variation.
Abb. 2 Landschaft der genomischen strukturellen Variation.
Die Autoren analysierten genomische strukturelle Variationen (SVs) bei indigenen und nicht-indigenen Australiern und stellten fest, dass die meisten SVs privat oder polymorph waren. Bemerkenswerterweise waren 48,5 % der SVs einzigartig für indigene Individuen, während 9,2 % einzigartig für nicht-indigene Individuen waren. Einzigartige SVs bei indigenen Individuen waren oft nicht annotiert und spezifisch für bestimmte Gemeinschaften. Genetische Unterschiede waren offensichtlich, mit ausgeprägten Clustern für verschiedene Gemeinschaften in der Hauptkoordinatenanalyse, und geteilte SVs unter indigenen Individuen waren selten, was ihre einzigartige genomische Vielfalt unterstreicht.
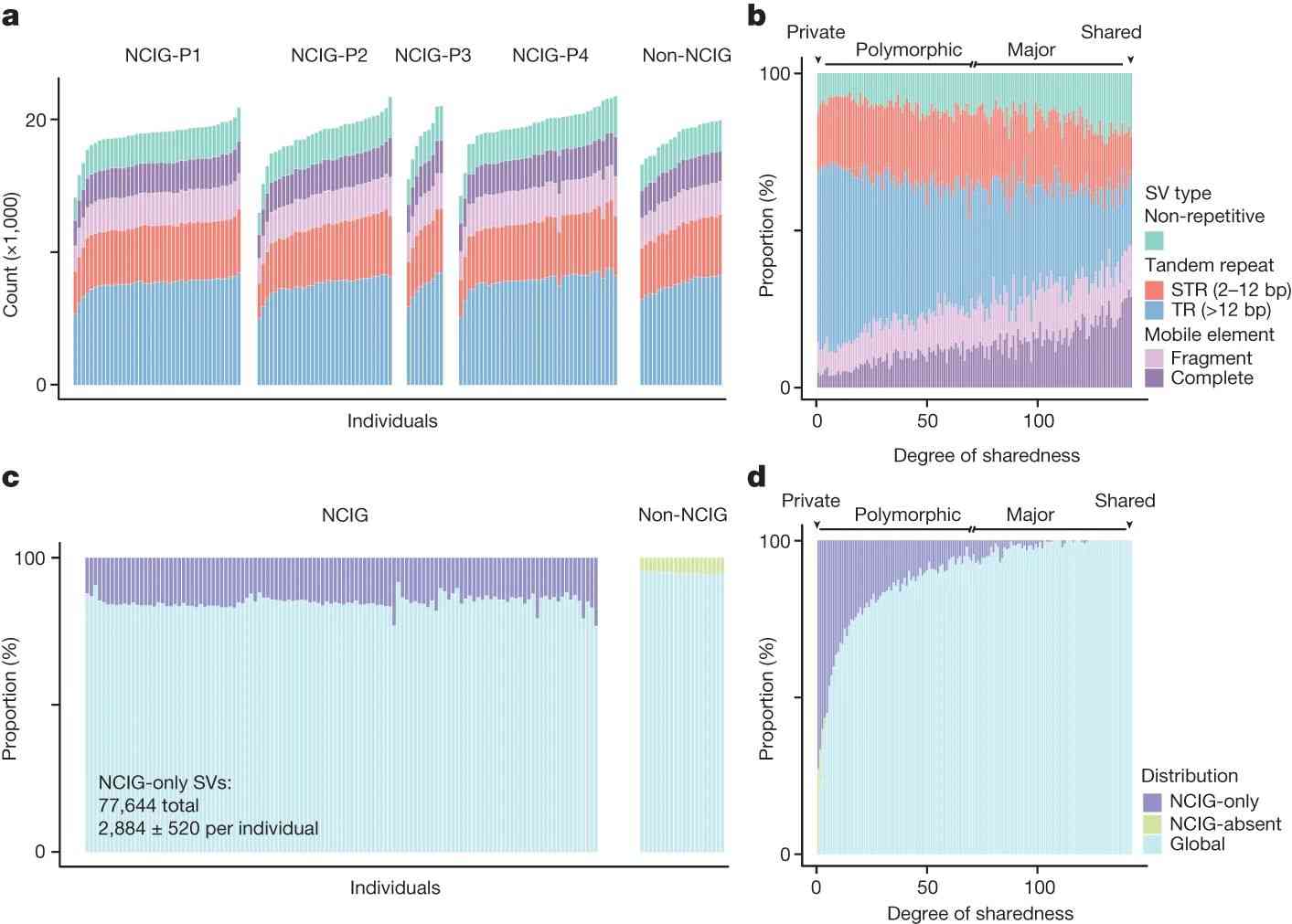 Abb. 3 Verteilung von SVs bei indigenen und nicht-indigenen Personen.
Abb. 3 Verteilung von SVs bei indigenen und nicht-indigenen Personen.
 Abb. 4 Verteilung der SVs zwischen indigenen Gemeinschaften.
Abb. 4 Verteilung der SVs zwischen indigenen Gemeinschaften.
Fazit
Diese Studie enthüllt die Landschaft der strukturellen Variationen der indigenen Australier durch Langsequenzierung und zeigt vielfältige Variationen, die dieser Population eigen sind. Dies unterstreicht die Notwendigkeit von ahnen-spezifischen Referenzdaten in der Genommedizin. Wir heben die genetische Heterogenität unter indigenen Gemeinschaften hervor und bieten Einblicke in breitere Muster der genomischen strukturellen Variation in menschlichen Populationen, insbesondere bei kurzen tandemwiederholungen (STRs), die Auswirkungen auf die Krankheitsdiagnose und -behandlung haben.
Referenz
- Reis, A.L.M., Rapadas, M., Hammond, J.M. et al. Die Landschaft der genomischen strukturellen Variation bei indigenen Australiern. Natur 624, 602–610 (2023).
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die unterschiedlichen Funktionen des Wildtyp- und R273H-Mutanten Δ133p53α regulieren unterschiedlich die Aggressivität von Glioblastomen und die durch Therapie induzierte Seneszenz.
Journal: Zellsterben & Krankheit
Jahr: 2024
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Generierung eines hoch attenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Journal: Mikrobielle Biotechnologie
Jahr: 2019
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genomanalysen und Replikationsstudien des afrikanischen Grünmeerkatzen-Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


