
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Fertige Bibliothekssequenzierung
Die Einführung von vorgefertigtem Bibliotheks-Sequenzieren
CD Genomics akzeptiert die von Kunden vorbereiteten Bibliotheken zur Sequenzierung. Sie können die Bibliotheken für einen vollständigen QC-Test einreichen und unseren Sequenzierungsdienst nutzen. Wir bieten die fortschrittlichsten und leistungsstärksten Sequenzierungsplattformen mit unterschiedlichen Kapazitäten und Lese-Längen, um jedem Projektumfang, Budget und Zeitrahmen gerecht zu werden. Unser QC-Bibliothek gewährleistet eine optimale Cluster-Generierung und maximale Datenausgabe für jeden Lauf. Verschiedene Proben können multiplexiert und zusammen sequenziert werden, solange sie die gleiche Lese-Länge haben. Unser hochqualifiziertes Team bietet Beratung zu den besten Sequenziergeräten für unterschiedliche Forschungsziele. Wir bieten auch KOSTENLOSE PhiX-Spike-Ins, Demultiplexing (FASTQ) und Datenübertragung über FTP an.
Fertige Bibliothekssequenzierung ist ein Hochdurchsatz-Sequenzierung Technik, die für die systematische Analyse des Genoms, Transkriptoms oder anderer Nukleinsäuresequenzen biologischer Proben verwendet wird. Sie umfasst die Vorbereitung von Nukleinsäuren aus der Probe in Bibliotheken, gefolgt von der Sequenzierung mit Hochdurchsatz-Sequenzierern, wodurch eine große Menge an Sequenzdaten für weitere Analysen generiert wird. Vorgefertigte Bibliotheken beinhalten das Fragmentieren der Nukleinsäuren der Probe vor der Sequenzierung und durch eine Reihe biochemischer Reaktionen das Einfügen von Adaptersequenzen, die mit der Sequenzierungsplattform kompatibel sind, wodurch sie für Hochdurchsatz-Sequenzierer geeignet gemacht werden.
Wenn Sie mehr über vorgefertigte Bibliothekssequenzierung erfahren möchten, können Sie auf unseren Artikel "Die Einführung und der Arbeitsablauf der vorgefertigten Bibliothekssequenzierung."
Vorteile von vorgefertigten Bibliothekssequenzierungen
- Hochdurchsatz: In der Lage, in kurzer Zeit eine große Menge an Sequenzdaten zu erzeugen.
- Vielseitigkeit: Anwendbar auf verschiedene Arten von Nukleinsäuremuster (DNA, RNA usw.).
- Flexibilität: Ermöglicht Forschung auf verschiedenen Ebenen wie Genomik, Transkriptomik, Epigenomikusw.
- Genauigkeit: Hochdurchsatz-Sequenzierung Die Technologie weist eine hohe Genauigkeit und Abdeckung auf.
- Zuverlässige Expertise: Ein kompetentes Team führt Sie durch jeden Schritt der Probenbearbeitung.
- Schnelle Bearbeitungszeit: Bietet die schnellste Bearbeitungszeit für die Probenverarbeitung, wodurch ein schnellerer Übergang von der Qualitätskontrolle zur Datenverbreitung ermöglicht wird und die Projektzeitpläne beschleunigt werden.
Anwendung von vorgefertigten Bibliothekssequenzierungen
- Genomik Forschung
- Transkriptomik Forschung
- Epigenetikforschung
- Mikrobiomforschung
- Onkologische Forschung
- Forschung zu seltenen Krankheiten
- Agrar- und Züchtungsforschung
- … und mehr
Vorgefertigter Bibliotheks-Sequenzierungs-Workflow
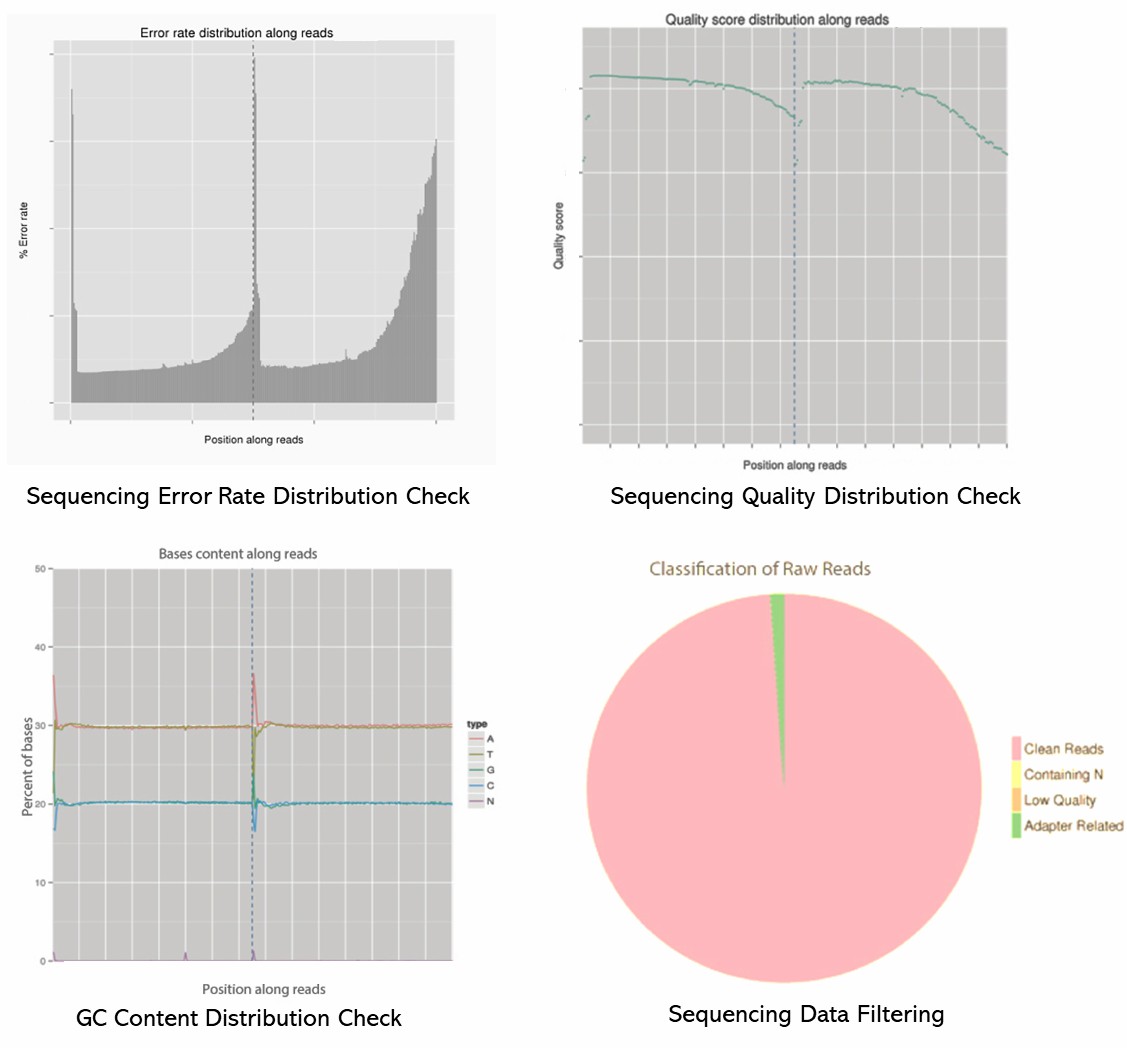
Die Qualitätsinspektionsmethode für die Größen und Konzentrationen der Bibliothek ist Qubit, Agilent Bioanalyzer.
Dienstspezifikationen
Beispielanforderungen
| Plattform | Mindestkonzentration | Datenmenge | Volumenanforderung |
|---|---|---|---|
| Novaseq-PE150 | 2 ng/μL | X<30G 30G≤X<100G 100G ≤ X ≤ 400G 400G < X < 800G 800G |
≥15 μL ≥25 μL ≥50 μL ≥70 μL ≥100 μL (zusätzliche 70 μL für eine weitere Bahn) |
| Nova- PE250 | 2 ng/μL | X<30GM 30M ≤ X < 100M 100M≤X<400M 400M |
≥15 μL ≥25 μL ≥50 μL ≥100 μL (zusätzliche 70 μL für eine weitere Bahn) |
| HiSeq-PE150 | 1 ng/μL | 1 Fahrspur | ≥10 μL |
| MiSeq-PE300 | 1 ng/μL | 1 Flusszelle | ≥10 μL |
Hinweis: Musterbeträge sind nur zur Referenz aufgeführt. Für detaillierte Informationen bitte Kontaktieren Sie uns mit Ihren maßgeschneiderten Anfragen.
Sequenzierungsstrategie
Illumina-Sequenzierung:
| Plattform | Lese-Länge (nt) | Einheit | Einheitsergebnis (rohe Cluster in Millionen) |
|---|---|---|---|
| NovaSeq 6000 Klicken |
PE50 | Gasse (SP) | 375-400 M |
| PE50 | Gasse (S1) | 650-800M | |
| PE50 | Gasse (S2) | 1.650-2.050 M | |
| PE100 | Lane (SP) | 375-400 M | |
| PE100 | Lane (S1) | 650-800M | |
| PE100 | Gasse (S2) | 1.650-2.050 M | |
| PE100 | Gasse (S4) | 4.000-5.000M | |
| PE150 | Gasse (SP) | 375-400 M | |
| PE150 | Lane (S1) | 650-800M | |
| PE150 | Gasse (S2) | 1.650-2.050 M | |
| PE150 | Lane (S4) | 4.000-5.000M | |
| PE250 | Gasse (SP) | 375-400 M | |
| HiSeq 4000 | SE50 | Gasse | 300-400 M |
| PE75 | Gasse | 300-400 M | |
| PE150 | Gasse | 300-400 M | |
| MiSeq | PE150 | Flowcell (Nano) | 1 M |
| PE250 | Flowcell (Nano) | 1 M | |
| PE150 | Flowcell (Mikro) | 4 M | |
| SE36 | Flowcell (V2) | 12-15 M | |
| PE25 | Flowcell (V2) | 12-15 M | |
| PE150 | Flowcell (V2) | 12-15 M | |
| PE250 | Flowcell (V2) | 12-15 M | |
| PE75 | Flowcell (V3i) | 22-25 M | |
| PE300 | Flowcell (V3) | 22-25 M |
| Plattform | Laufart |
|---|---|
| Pacbio Sequl II SMRT-Zelle 8M | HiFi-Sequenzierung |
| CLR-Sequenzierung |
Bioinformatische Analyse
- Datenqualitätskontrolle
- Datenvorverarbeitung
- Ausrichtung
- Versammlung
- Variantenerkennung
- Genexpressionsanalyse
- Funktionale Annotation
- … und mehr
Hinweis: Die empfohlenen Datenoutputs und Analyseinhalte, die angezeigt werden, dienen nur zur Referenz. Für detaillierte Informationen, bitte Kontaktieren Sie uns mit Ihren maßgeschneiderten Anfragen.
Analyse-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details in der vorgefertigten Bibliothekssequenzierung für Ihr Schreiben (Anpassung)
Demo-Ergebnisse
Häufig gestellte Fragen zur vorgefertigten Bibliothek Seq
1. Welche Arten von Proben können für die Sequenzierung von vorgefertigten Bibliotheken verwendet werden?
Für die Sequenzierung mit vorgefertigten Bibliotheken werden verschiedene Probenarten verwendet, darunter:
- Genomisches DNA: Verwendet für Whole-Genome-Sequenzierung (WGS) oder gezielte Sequenzierung Anwendungen.
- RNA: Eingesetzt in der Transkriptom-SequenzierungRNA-Seq), die totale RNA, mRNA und kleine RNA umfasst.
- cDNA: Wird verwendet, um revers-transkribierte RNA in bestimmten Kontexten zu sequenzieren.
2. Wie wird die Qualität der vorgefertigten Bibliothek bewertet?
Die Qualität der vorgefertigten Bibliothek wird bewertet durch:
- Größenverteilung: Analyse der Fragmentgrößenverteilung mit einem Instrument wie dem Agilent Bioanalyzer oder TapeStation.
- Konzentrationsmessung: Quantifizierung der Bibliothekskonzentration mit einem Qubit-Fluorometer oder quantitativer PCR (qPCR).
- Qualitätskontrollmetriken: Überprüfung auf Adapter-Dimere, Kontamination und allgemeine Qualität mithilfe von Tools wie FastQC für rohe Sequierungsdaten.
3. Worauf bezieht sich die Einheitlichkeit der Bibliotheksausgaben?
Während der Sequenzierung bleibt das Ausgabe-Datenvolumen von Bibliotheken im gleichen Lane einheitlich im Verhältnis zur Menge des Bibliotheksinputs. Wenn beispielsweise 5 Bibliotheken gemischt werden und insgesamt 50G Daten erzielt werden, sollte es nicht vorkommen, dass eine Bibliothek 30G Daten liefert, während eine andere nur 5G liefert; jede Bibliothek sollte idealerweise gleichmäßig mit 10G beitragen.
Kann die Verwendung von vorgefertigten Bibliothekssequenzen für die Einzelzellanalyse genutzt werden?
Natürlich kann die vorgefertigte Bibliothekssequenzierung für die Einzelzellanalyse umgenutzt werden. Techniken wie die Einzelzell-RNA-Sequenzierung (scRNA-seq) beinhalten das Isolieren einzelner Zellen, das Rücktranskribieren von RNA in cDNA und das Erstellen von Sequenzierungsbibliotheken aus der resultierenden Einzelzell-cDNA. Diese Methodik ermöglicht die Untersuchung der Genexpression auf Einzelzellebene und eröffnet Einblicke in die zelluläre Heterogenität und Funktion.
Fertige Bibliothek Seq Fallstudien
Landschaft somatischer Mutationen in 560 gesamten Genomsequenzen von Brustkrebs
Zeitschrift: Natur
Impactfaktor: 64,8001
Veröffentlicht: 2. Mai 2016
Hintergrund
Die mutationale Theorie des Krebses postuliert, dass spezifische Veränderungen der DNA-Sequenz, die als "Treiber"-Mutationen bezeichnet werden, den Zellen proliferative Vorteile verleihen und somit das Auftreten maligner Zellpopulationen fördern. Diese Mutationen können aus verschiedenen Mechanismen resultieren, einschließlich der Exposition gegenüber Mutagenen, Fehlern in den DNA-Reparaturmechanismen und Ungenauigkeiten während der DNA-Replikation. Technologischer Fortschritt, der von der Karyotypanalyse bis hin zu Hochdurchsatz- DNA-Sequenzierung, hat die Abgrenzung von krebsassoziierten Mutationen erheblich verbessert. Dennoch wurde die Hauptaufmerksamkeit auf protein-codierende Regionen gerichtet, wodurch wichtige Fragen zu Mutationen in nicht-codierenden Regionen und den zugrunde liegenden mutagenen Prozessen beim Brustkrebs unbeantwortet blieben. Um diese Wissenslücken zu schließen, führten wir eine eingehende Analyse von Whole-Genome-Sequenzen von 560 Brustkrebsfällen, die auf eine umfassende Aufklärung somatischer Mutationen in diesem Kontext abzielen.
Methoden
- 560 Brustkrebserkrankungen
- Normales Gewebe
- DNA-Extraktion
- Gesamte RNA-Extraktion
- Kurze Inserts von 500 bp genomischen Bibliotheken
- 350bp poly-A-selektierte transkriptionelle Bibliotheken
- Hochdurchsatz-Sequenzierung
- Ausrichtung
- Verarbeitung von genomischen Daten
- Identifizierung neuartiger Brustkrebs-Gene
- Analyse von Mutationssignaturen
- Umgruppierungssignaturen
- Einzelne Patienten-Ganzgenomprofile
Ergebnisse
Die gesamten Genome von 560 Brustkrebserkrankungen und passenden nicht-neoplastischen Geweben wurden sequenziert, wobei zahlreiche somatische Mutationen entdeckt wurden. Durch die Kombination von Daten aus verschiedenen Quellen wurden neue Krebs-Gene identifiziert und Treibermutationen definiert. Genomische Umstellungen und Veränderungen der Kopienzahl trugen weiter zur Identifizierung von Treibermutationen bei, wobei TP53, PIK3CA und MYC zu den am häufigsten mutierten Genen gehören.
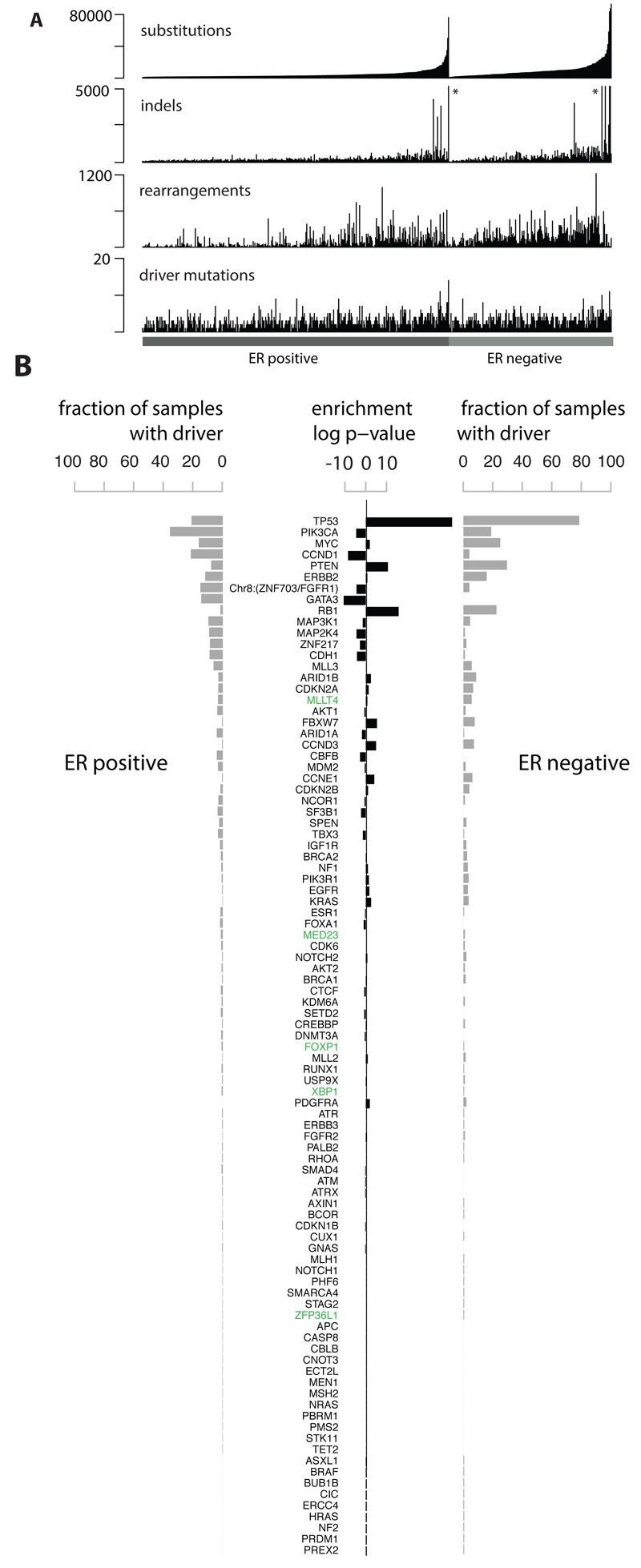 Abb. 1. Kohorte und Katalog somatischer Mutationen in 560 Brustkrebserkrankungen.
Abb. 1. Kohorte und Katalog somatischer Mutationen in 560 Brustkrebserkrankungen.
Wir haben nicht-kodierende somatische Mutationen untersucht und wiederkehrende Mutationen im Promotor von PLEKHS1 identifiziert, die mit den mutationalen Signaturen 2 und 13 verknüpft sind. Ähnliche Mutationen wurden in den Promotoren von TBC1D12 und WDR74 festgestellt, was auf eine potenzielle Hypermutabilität hindeutet. Darüber hinaus wurden Mutationen in den langen nicht-kodierenden RNAs MALAT1 und NEAT1 entdeckt, deren Bedeutung als Treibermutationen jedoch ungewiss bleibt.
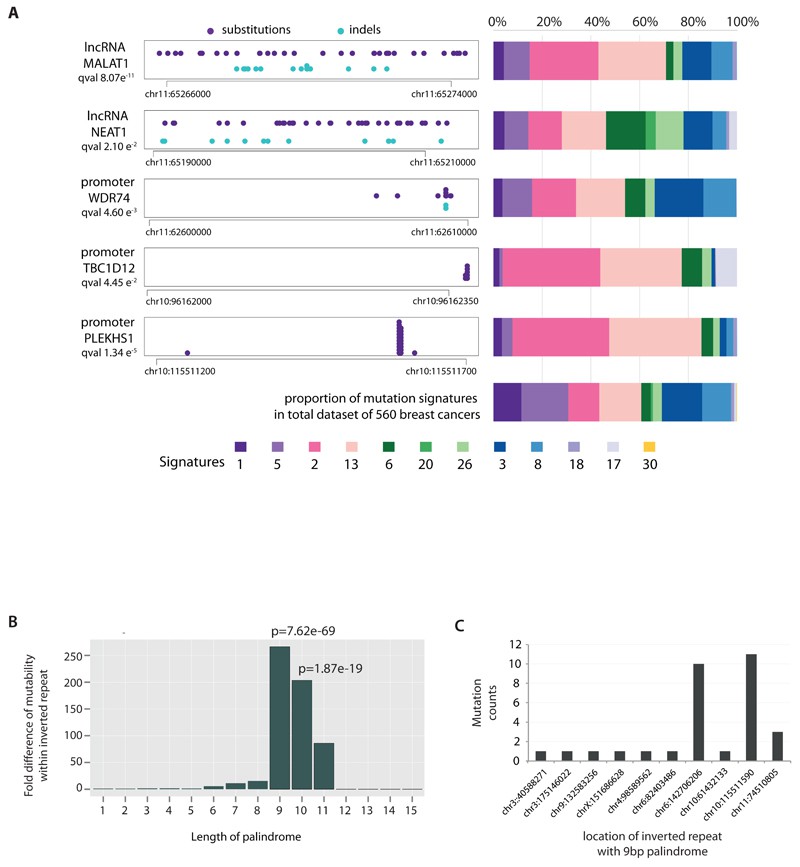 Abb. 2. Nicht-kodierende Analysen von Brustkrebsgenomen.
Abb. 2. Nicht-kodierende Analysen von Brustkrebsgenomen.
Fazit
Der Fortschritt zu einem umfassenden Verständnis der Genetik von Brustkrebs ist im Gange und zeigt mehrere mutationale Signaturen sowie die Beteiligung zahlreicher Krebs-Gene. Die Seltenheit von dominant wirkenden Fusionsgenen und nicht-kodierenden Treibermutationen deutet jedoch auf zusätzliche Komplexitäten hin. Dennoch bleiben viele Fragen offen, einschließlich der Identifizierung weiterer Krebs-Gene und der Rollen von Viren oder Mikroben. Eine weitere Untersuchung der gesamten Genomsequenzen von Brustkrebspatienten ist erforderlich, um die somatische mutationale Landschaft der Krankheit vollständig zu erhellen.
Referenz:
- Nik-Zainal S, Davies H, Staaf J, et al. Landschaft somatischer Mutationen in 560 Ganzgenomsequenzen von Brustkrebs. Natur, 2016, 534(7605): 47-54.
Verwandte Veröffentlichungen
Hier sind einige Veröffentlichungen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Die HLA-Klasse-I-Immunopeptidome der AAV-Kapsidproteine
Zeitschrift: Frontiers in Immunologie
Jahr: 2023
Isolation und Charakterisierung neuer menschlicher Trägerpeptide aus zwei wichtigen Impfstoff-Immunogenen
Zeitschrift: Impfstoff
Jahr: 2020
Änderung von Gewicht, BMI und Körperzusammensetzung in einer bevölkerungsbasierten Intervention im Vergleich zu einer genetisch basierten Intervention: Die NOW-Studie
Zeitschrift: Fettleibigkeit
Jahr: 2020
Sarecyclin hemmt die Proteintranslation im Cutibacterium acnes 70S-Ribosom durch einen Zwei-Stellen-Mechanismus.
Zeitschrift: Nucleic Acids Research
Jahr: 2023
Identifizierung eines Darmkommensalen, der die blutdrucksenkende Wirkung von Ester-Angiotensin-Converting-Enzym-Hemmern beeinträchtigt.
Journal: Hypertonie
Jahr: 2022
Eine Splice-Variante im SLC16A8-Gen führt zu einem Defizit beim Laktattransport in aus menschlichen iPS-Zellen abgeleiteten retinalen Pigmentepithelzellen.
Journal: Zellen
Jahr: 2021
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


