
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Vollständige 16S/18S/ITS Amplicon-Sequenzierung
In Bezug auf die umfangreiche Erfahrung in der Sequenzierung ist CD Genomics stolz darauf, unseren Kunden vollständige 16S/18S/ITS rRNA-Dienstleistungen mit der besten Qualität und dem wettbewerbsfähigsten Preis anzubieten.
Was ist die vollständige 16S/18S/ITS-Amplikon-Sequenzierung?
Das 16S rRNA-Gen ist zwischen verschiedenen Arten von Bakterien und Archaeen stark konserviert und enthält neun hypervariable Regionen (V1-V9), die etwa 30 bis 100 Basenpaare lang sind und zwischen Bakterien stark variieren. Stark konservierte Regionen ermöglichen es Forschern, Primerpaare zu entwerfen, die die hypervariable Region des 16S-Gens ihrer Wahl genau und zuverlässig amplifizieren, um die Identifizierung oder Charakterisierung verschiedener bakterieller Gemeinschaften zu erreichen.
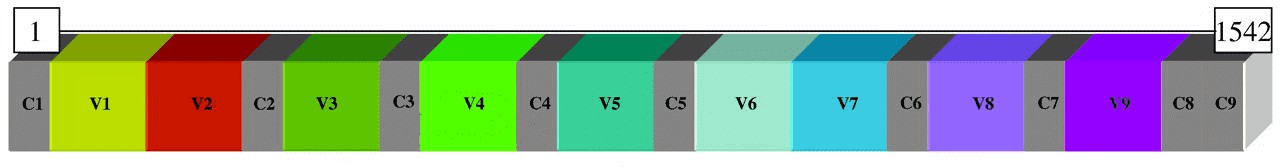
Ähnlich wie die bakteriellen 16S rRNA-Gene weist das eukaryotische 18S rRNA-Gen konservierte und variable Regionen auf. 18S rRNA-Gen-Sequenzen und ihre zugehörigen transkribierten Spacer (interne transkribierte Spacer; ITS) werden verwendet, um Pilze und Eukaryoten zu klassifizieren.
Basierend auf der Entwicklung der Sequenzierungstechnologie, 16S/18S/ITS rRNA-Gen-Sequenzierung ist das beste Werkzeug, um bakterielle und pilzliche Taxonomie sowie molekulare Phylogenie zu studieren. Indem es die Vorteile von PacBio SMRT-Langsequenzierung Technologie, CD Genomics kann einen umfassenden 16S/18S/ITS rRNA-Sequenzierungsdienst anbieten, um Ihre Forschung besser zu unterstützen.
Die Vollsequenzierung von 16S/18S/ITS umfasst die Extraktion von mikrobieller DNA aus einer gegebenen Probe, gefolgt von der Amplifikation des mikrobiellen 16S rDNA, 18S rDNA oder des ITS-Bereichs in seiner Gesamtheit unter Verwendung universeller Primer und anschließender Sequenzierung. Diese Methodik erhöht nicht nur die Präzision der Artenidentifikation dank verbesserter Auflösung, sondern verbessert auch die Genauigkeit bei der Entschlüsselung der mikrobiellen Zusammensetzung innerhalb der Proben. Folglich bietet dies einen integrativeren Einblick in die Struktur der mikrobiellen Gemeinschaft.
Mit einer so erhöhten Auflösung verschiebt sich der Fokus natürlich auf Studien auf Artenebene. Dies stellt einen bedeutenden Fortschritt im Vergleich zu früheren Studien der zweiten Generation dar, die sich auf das Genus- und Artenniveau unter Verwendung von 16S konzentrierten. Vollständige 16S-Daten der dritten Generation können umfassendere und komplexere Analysen auf Stammebene bieten, wodurch die Gesamtergebnisse der Forschung relevanter für die ökologische Funktionalität werden. Dieser Ansatz hat erhebliche Auswirkungen auf die Multi-Omics-Korrelationen sowie auf nachfolgende experimentelle Richtlinien und Validierungen, was ihm eine bedeutende Bedeutung verleiht. Aus der Perspektive der Multi-Omics-Korrelation zeigen feinere Daten oft klarere lokale Muster und umfassen viele Details, die zuvor entweder übersehen oder unzugänglich waren.
Vorteile der Voll-Längen 16S/18S/ITS Amplicon-Sequenzierung
- Längere Texte für erhöhte Genauigkeit.
- Höhere Auflösung und Präzision bei der Artenidentifikation.
- Genauere Rekonstruktion von mikrobiellen Gemeinschaften.
- Reife Sequenzierungsplattform: PacBio SMRT System.
- Hochwertige Datenqualitätssicherung.
- Genaues Identifizieren mit mehreren Optionen für CCS-Lesungen.
- Reiche Projekterfahrung.
- Vielfältige Analyseinhalte.
- Kurze Zyklen mit wettbewerbsfähigen Preisvorteilen.
Anwendungen der Voll-Längen 16S/18S/ITS Amplicon-Sequenzierung
- Forschung über die Beziehung zwischen häufigen Krankheiten und der menschlichen Mikrobiota.
- Tierbereich: Studien zur Beziehung zwischen dem Darm, dem Pansen (wie methanproduzierenden Mikrobengemeinschaften) und der Gesundheit/Nährstoffverdauung von Tieren.
- Landwirtschaftlich Feld: Forschung zu Rhizosphären-Mikrobiota und Pflanzeninteraktionen sowie landwirtschaftlichen Anbau-/Düngemethoden und Bodenmikroben-Gemeinschaften.
- Umwelt- Feld: Untersuchungen zur Behandlung von Dunst, Abwasserbewirtschaftung, Abbau von Erdöl, Behandlung von saurem Bergbauabfluss und Forschung zur marinen Umwelt.
- Spezielle extreme Umgebungen: Forschung zu mikrobiellen Gemeinschaften unter extremen Umweltbedingungen.
Workflow für die Voll-Längen 16S/18S/ITS Amplicon-Sequenzierung
Dienstspezifikation
Beispielanforderungen:
|
|
 |
Sequenzierung:
|
 |
Datenanalyse
|
Bioinformatik-Pipeline

Liefergegenstände
- Die ursprünglichen Sequenzierungsdaten
- Experimentelle Ergebnisse
- Datenanalysebericht
- Details zur Vollständigen 16S/18S/ITS Amplicon-Sequenzierung für Ihr Schreiben (Anpassung)
Referenzen:
- Callahan B J, Wong J, Heiner C, et al. Hochdurchsatz-Amplicon-Sequenzierung des vollständigen 16S rRNA-Gens mit Einzel-Nukleotid-Auflösung. Nukleinsäureforschung, 2019, 47(18): e103-e103.
- Lam T Y C, Mei R, Wu Z, et al. Überlegene Auflösungscharakterisierung der mikrobiellen Vielfalt in anaeroben Vergärern durch die Sequenzierung von Voll-Längen 16S rRNA-Gen-Amplikonen. Wasserforschung, 2020, 178: 115815.
- Hui M, Wang A, Cheng J, et al. Die vollständige Sequenzierung von 16S rRNA-Amplikonen zeigt die Variation der epibiontischen Mikrobiota, die mit zwei Garnelenarten der Familie Alvinocarididae assoziiert ist: möglicherweise co-bestimmt durch Umweltheterogenität und spezifische Erkennung der Wirte. PeerJ, 2022, 10: e13758.
Demonstrationsergebnisse
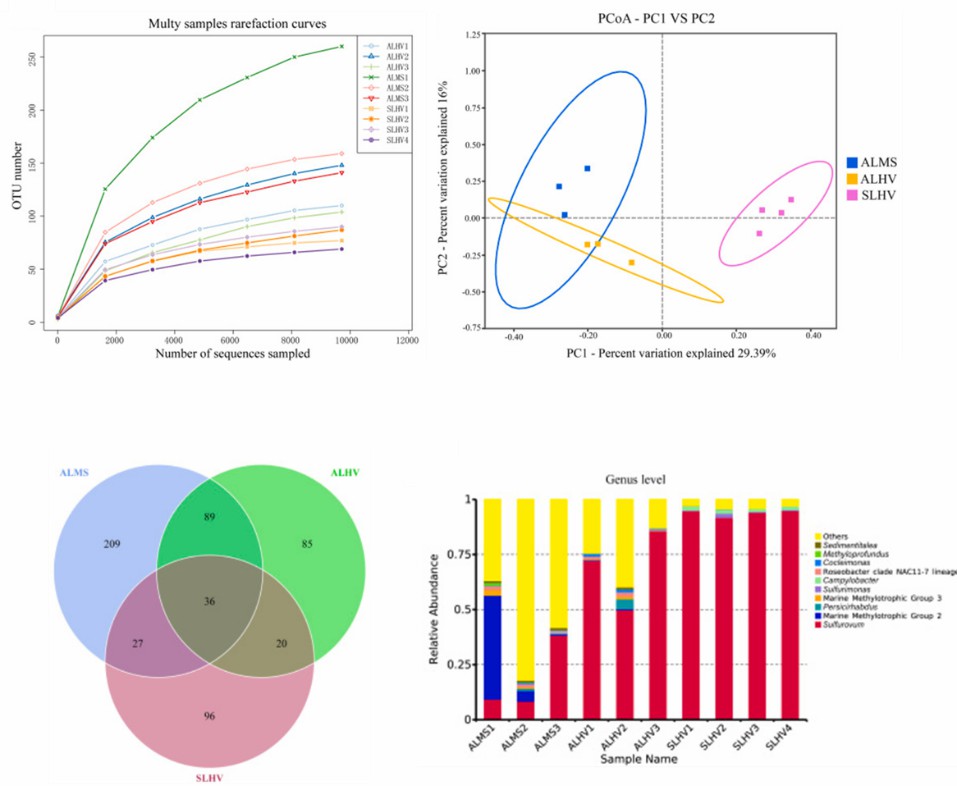 Verwendung der Ergebnisse der 16S rRNA-Amplikon-Sequenzierung zur Darstellung. (Hui et al., 2022)
Verwendung der Ergebnisse der 16S rRNA-Amplikon-Sequenzierung zur Darstellung. (Hui et al., 2022)
Häufig gestellte Fragen zu Full-Length 16S/18S/ITS Amplicon-Sequenzierungen
1. Was sind die Vorteile der Amplicon-Sequenzierung der dritten Generation?
Die Amplicon-Sequenzierung der dritten Generation bietet mehrere Vorteile. Durch die Nutzung der PacBio SMRT Einzelmolekül-Sequenzierung Technologie, sie bietet ultra-lange Reads, die die Abdeckung von vollständigen ribosomalen kleinen Untereinheiten 16S/18S/ITS-Regionen ermöglichen. Dies adressiert die technische Herausforderung von Short-Read-Produkten, die auf die Analyse nur lokaler variabler Regionen beschränkt sind. Darüber hinaus ermöglicht sie eine wirklich präzise mikrobielle Klassifikation und Identifizierung zu angemessenen Kosten.
2. Warum sich für die vollständige mikrobielle Vielfalt entscheiden?
Die Sequenzierungstechnik der zweiten Generation analysiert hauptsächlich partielle Sequenzen des 16S rRNA-Gens. Verschiedene Forscher analysieren jedoch unterschiedliche Regionen der 16S rRNA-Gene in verschiedenen Bakterien, und die Wahl der PCR-Primer für kurze Amplifikate aus verschiedenen hypervariablen Regionen kann die abgeleitete Genauigkeit der Gemeinschaft und die Empfindlichkeit gegenüber bestimmten bakteriellen Gruppen beeinflussen. Diese Situation macht Vergleiche auf globaler Ebene in Mikrobiomstudien herausfordernd. Darüber hinaus kann aufgrund der begrenzten Mutationsinformationen, die einige hypervariable Regionen tragen, keine Artenebene-Annotation erreicht werden. Daher wird die Diversität von Mikrobiomen der zweiten Generation typischerweise auf Gattungsebene und höher untersucht. Im Gegensatz dazu kann die Sequenzierung der dritten Generation alle hypervariablen Regionen auf einmal messen. Diese Fähigkeit beseitigt die Präferenzen, die durch unterschiedliche Primer entstehen, und bietet eine neuartige Methode zur Verbesserung der taxonomischen Auflösung mikrobieller Gemeinschaften.
3. Wie identifiziert man Bakterienarten?
Vergleichen Sie die erhaltenen 16S rDNA-Sequenzen mit Referenz-16S rDNA-Sequenzen in GenBank oder Eztaxon. Bei mehr als 97 % Sequenzähnlichkeit können sie als dieselbe Bakterienart betrachtet werden. Für eine genauere Identifizierung sollten DNA-Hybridisierung, genomischer GC-Gehalt sowie physiologische und biochemische Indizes verwendet werden.
4. Die Anforderungen an die Proben.
Wenn Sie kultivierte Mikroben einreichen, stellen Sie bitte sicher, dass sie mindestens dreimal gereinigt wurden. Geben Sie außerdem eine klare Angabe zu den Kulturbedingungen. Falls ein spezielles Medium benötigt wird, stellen Sie dies bitte zur Verfügung.
Wenn Sie genomische DNA-Proben einreichen, sind mehr als 500 ng erforderlich.
Für grampositive Mikroben reichen Sie bitte genomische DNA-Proben ein.
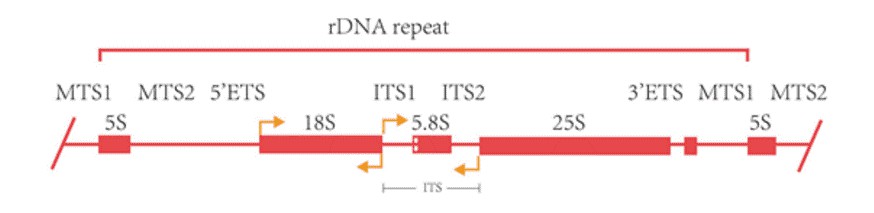
5. Wie lang ist die vollständige Länge der 16S/18S/ITS-Sequenz?
Das 16S rRNA-Gen prokaryotischer Mikroben hat eine Länge von etwa 1500 bp und umfasst zehn konservierte Regionen sowie neun hochvariable Regionen (V1-V9). Es weist sowohl ein hohes Maß an Konservierung als auch Spezifität auf, wobei die konservierten Regionen phylogenetische Beziehungen zwischen Gattungen widerspiegeln und die variablen Regionen die Variation zwischen den Gattungen darstellen. Das 18S rRNA-Gen eukaryotischer Mikroben hat eine Sequenzlänge von 1500-2000 bp und weist ebenfalls sowohl konservierte als auch variable Regionen auf. Aufgrund seiner konservativen Evolutionsrate ist es für die taxonomische Identifizierung auf Artenebene und darüber hinaus geeignet. Der interne transkribierte Spacer (ITS) eukaryotischer Mikroben, der etwa 500 bp lang ist, behält eine hohe Konservierung zwischen verschiedenen Stämmen innerhalb einer Art bei, weist jedoch erhebliche Variation zwischen den Arten auf. Dies macht ihn extrem polychromatisch und beweist seine breite Anwendung in phylogenetischen Studien und Forschungen zwischen Arten.
Vollständige 16S/18S/ITS Amplicon-Sequenzierungs-Fallstudien
Analyse des Mikrobioms des Mausdarms mittels Voll-Längen 16S rRNA Amplicon-Sequenzierung
Journal: Wissenschaftliche Berichte
Impactfaktor: 4,6
Veröffentlicht: 14. Juli 2016
Zusammenfassung
Das Aufkommen von Next-Generation-Sequencing-Technologien hat die detaillierte Klärung der Zusammensetzung mikrobieller Gemeinschaften unterstützt und bietet im Vergleich zu früheren Methoden eine verbesserte Genauigkeit und Durchsatz. Die Autoren nutzten Nanoporen-Sequenzierungsplattformen, um eine Bibliothek vollständiger 16S rRNA-Amplikons zu sequenzieren, die aus dem Mikrobiom des Darms von Mäusen vorbereitet wurde. Auf der Art-Ebene übertrifft die Nanoporen-Sequenzierung die Kurzlesesequenzierung bei der Identifizierung von mehr Arten und trägt somit zu einer genauen Charakterisierung der bakteriellen Gemeinschaftszusammensetzung bei.
Methoden
- Mikrobiell Metagenomik DNA
- Vollständige 16S rDNA Amplicon-Sequenzierung
- Illumina-Sequenzierung
- OTU-Taxonomie
- Sequenzierungsgenauigkeit berechnet
Ergebnisse
1. Statistischer Vergleich zwischen Short-Read- und Nanopore-Sequenzierungsdaten
Das Dataset der Kurzlesesequenzierung wurde unter Verwendung des Ansatzes der Operational Taxonomic Units (OTU) analysiert. Andererseits wurde die Zusammensetzung der mikrobiellen Gemeinschaft basierend auf den durch eine Systematisierungsmethode gewonnenen Nanopore-Sequenzierungsdaten bestimmt, die als taxonomisch überwachte Analyse bekannt ist und Sequenzen direkt basierend auf der Sequenzähnlichkeit in taxonomische Kategorien einordnet. Trotz des vernachlässigbaren Niveaus der stichproben-spezifischen Phylogenie, das beobachtet wurde, und der relativ hohen Fehlerquote (20,4%), die aus den Nanopore-Sequenzierungsdaten identifiziert wurde, wurde eine hohe Reproduzierbarkeit und Korrelation zwischen biologischen Replikaten erreicht. Folglich ermöglicht die Anwendung der Nanopore-Amplikon-Sequenzierung die wiederholte Erfassung von 34 Hauptsystemtypen.
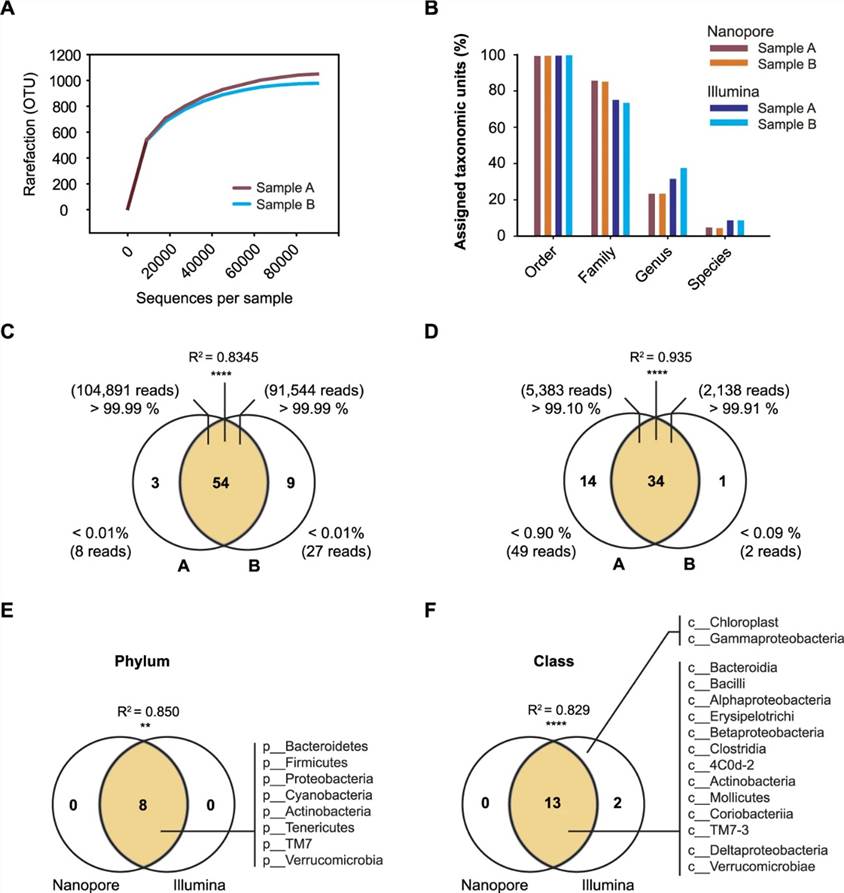 Abbildung 1. Statistischer Vergleich zwischen Kurzlese- und Nanopore-Sequenzierungsdaten.
Abbildung 1. Statistischer Vergleich zwischen Kurzlese- und Nanopore-Sequenzierungsdaten.
2. Vergleich der mikrobiellen Zusammensetzung, die durch zwei Sequenzierungsplattformen bestimmt wurde
Die Autoren verglichen die mikrobielle Zusammensetzung, die mit zwei Sequenzierungsplattformen bestimmt wurde. Beide Plattformen identifizierten 8 bakterielle Phyla und 13 bakterielle Klassen (wie dargestellt). Es gab eine bemerkenswerte statistische Ähnlichkeit zwischen der Kurzlese- und der Nanopore-Sequenzierung hinsichtlich der relativen Anteile und Klassen der Hauptphyla. Im Allgemeinen wurden signifikante Ähnlichkeiten in der bakteriellen Zusammensetzung zwischen den beiden Plattformen auf der Ordnung-, Familien- und Artenebene beobachtet. Aus der vergleichenden Analyse der mikrobiellen Zusammensetzung, die unabhängig über Nanopore- und Kurzlesesequenzierungsplattformen analysiert wurde, geht hervor, dass die Nanopore-Sequenzierung die mikrobielle Zusammensetzung auf Artenebene genau identifizieren kann.
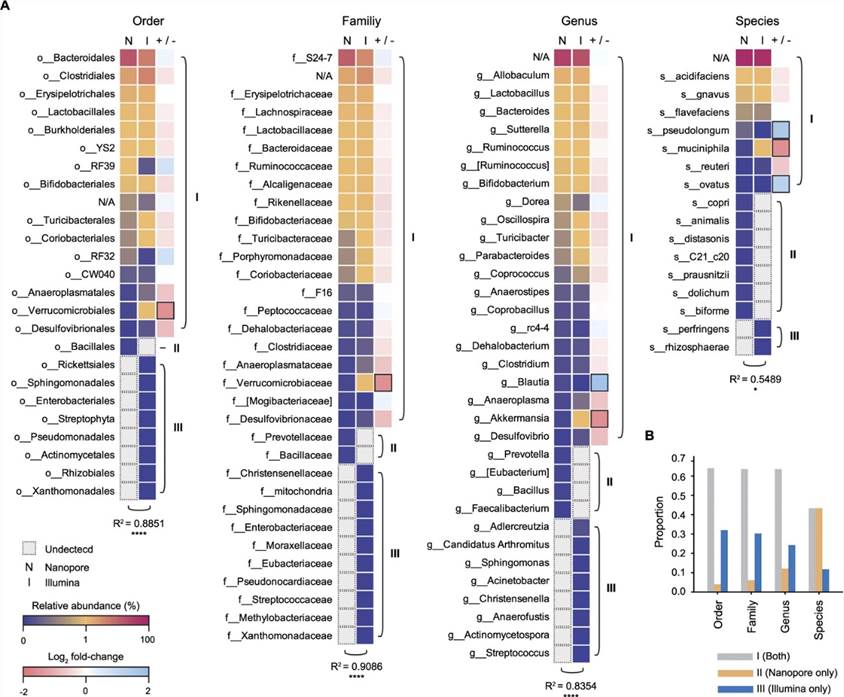 Abbildung 2. Vergleich der Mikrobiota-Zusammensetzungen im Mausdarm zwischen zwei Sequenzierungsplattformen bei tiefergehenden Klassifikationen (Ordnung bis Art).
Abbildung 2. Vergleich der Mikrobiota-Zusammensetzungen im Mausdarm zwischen zwei Sequenzierungsplattformen bei tiefergehenden Klassifikationen (Ordnung bis Art).
3. Speziesdetektion mittels Voll-Längen 16S rDNA Amplicon-Sequenzierung
In Bezug auf die taxonomische Auflösung vom Phylum bis zur Speziesebene sind die relativen Anteile von Gruppe I (beide Sequenzierungsplattformen) und Gruppe III (nur Kurzlesesequenzierung) gesunken, während Gruppe II (nur Nanopore-Sequenzierung) einen Anstieg verzeichnet hat. In diesem Zusammenhang postulieren wir, dass die langen Reads, die durch Nanopore-Sequenzierung erzeugt werden, mehrere hypervariable Regionen überqueren und aufgrund ihrer nahezu vollständigen 16S rDNA-Reads möglicherweise die Illumina-Sequenzierung bei der Unterscheidung der mikrobiellen Gemeinschaftsstruktur übertreffen. Um die Auswirkungen längerer Reads auf die Analyse der mikrobiellen Gemeinschaft auf Speziesebene zu bestimmen, führten wir eine phylogenetische Analyse des zusammengesetzten Datensatzes basierend auf Priorität durch und identifizierten 16 phylogenetisch unterschiedliche Arten, die über 13 Gattungen verteilt sind (wie dargestellt).
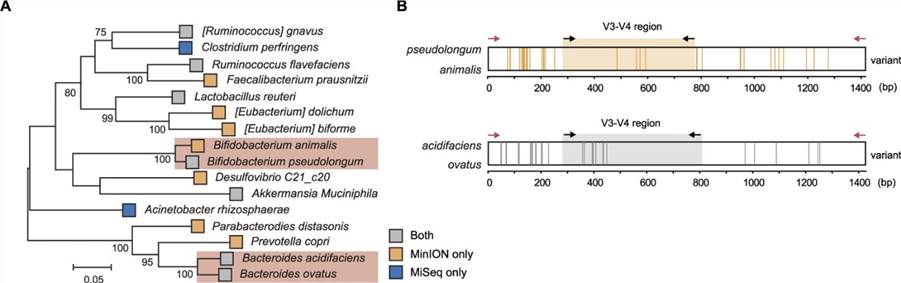 Abbildung 3. Phylogenetische Analyse der Mikrobiota im Mausdarm.
Abbildung 3. Phylogenetische Analyse der Mikrobiota im Mausdarm.
Fazit
Die Autoren untersuchten das Potenzial des Sequenziergeräts der dritten Generation, MinION, zur Identifizierung der mikrobiellen Gemeinschaftszusammensetzung in Stuhlproben von Mäusen. Durch den Einsatz von Nanoporen-Sequenzierung für die vollständige 16S rRNA-Amplicon-Sequenzierung konnten sie schnell, genau und effizient die mikrobielle Vielfalt auf Artenebene bestimmen. Der Erfolg dieser Studie bei der Klärung der mikrobiellen Gemeinschaftszusammensetzung deutet auf das zukünftige Potenzial der Methode hin. Die Autoren erwarten, dass dieser Ansatz die Anwendbarkeit von Analysen mikrobieller Gemeinschaften in biologischen, klinischen und umweltbezogenen Bereichen erweitern wird.
Referenz:
- Shin J, Lee S, Go M J, et al. Analyse des Mikrobioms des Mausdarms mittels vollständiger 16S rRNA-Amplikon-Sequenzierung. Wissenschaftliche Berichte, 2016, 6(1): 29681.
Verwandte Veröffentlichungen
Hier sind einige Publikationen, die erfolgreich mit unseren Dienstleistungen oder anderen verwandten Dienstleistungen veröffentlicht wurden:
Bakterielle Gemeinschaften von Cassiopea in den Florida Keys teilen sich wichtige bakterielle Taxa mit Korallen-Mikrobiomen.
Journal: bioRxiv
Jahr: 2024
Produktion eines Bakteriocin-ähnlichen Proteins PEG 446 aus Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotika und antimikrobielle Proteine
Jahr: 2024
Entwirrung der Rolle von Pathobionten aus Bacteroides-Arten bei entzündlichen Darmerkrankungen
Journal: bioRxiv
Jahr: 2023
Eine Chromosomenebene-Genomressource zur Untersuchung von Virulenzmechanismen und der Evolution des Kaffeerostpathogens Hemileia vastatrix
Journal: bioRxiv
Jahr: 2022
Streptomyces buecherae sp. nov., ein Actinobacterium, das aus mehreren Fledermausarten isoliert wurde
Journal: Antonie van Leeuwenhoek
Jahr: 2020
Mehr anzeigen Artikel, die von unseren Kunden veröffentlicht wurden.


