
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Was ist De Novo Whole Genome Sequencing?
Die de-novo-Sequenzierung des gesamten Genoms ist der Prozess, ein vollständiges Genom von Grund auf neu zusammenzustellen – ohne auf eine bereits vorhandene Referenzsequenz zurückzugreifen. Im Gegensatz zur Resequenzierung, bei der Reads an ein bekanntes Genom ausgerichtet werden, rekonstruiert die de-novo-Assemblierung das Genom direkt aus Sequenzfragmenten, was sie zur bevorzugten Methode für Arten macht, die kein Referenzgenom haben, oder für Projekte, die eine unvoreingenommene Sicht auf den genomischen Inhalt erfordern.
CD Genomics bietet umfassende de novo Genomsequenzierungsdienste, die das gesamte Spektrum biologischer Komplexität abdecken – von viralen, bakteriellen und pilzlichen Genomen bis hin zu großen Pflanzen- und Tiergenomen. Unser Multi-Plattform-Ansatz integriert Illumina-Kurzlesungen, PacBio HiFi-Lesungen, Oxford Nanopore-Ultra-Lesungen und Hi-C-Chromatin-Interaktionsdaten, um Assemblierungen zu liefern, die von standardmäßiger Entwurfqualität bis hin zu telomer-zu-telomer (T2T) lückenloser Auflösung reichen, einschließlich der Konstruktion von Pan-Genomen, wenn eine Diversität auf Bevölkerungsebene erforderlich ist.
De Novo Sequenzierung über alle Organismustypen hinweg
De-novo-Genomsequenzierung ist kein Einheitsdienst. Verschiedene Organismusgruppen stellen unterschiedliche Herausforderungen in Bezug auf Genomgröße, Ploidie, Wiederholungsinhalt und Heterozygotie dar. Unser Dienstleistungsportfolio ist so strukturiert, dass es diese Unterschiede mit maßgeschneiderten experimentellen und analytischen Strategien anspricht.
Mikrobielle Whole-Genome-De-Novo-Sequenzierung (Viral / Bakteriell / Pilz)
Mikrobielle Genome reichen von wenigen Kilobasen (Viren) bis zu mehreren Megabasen (Pilze). Unser mikrobielles De-novo-Workflow kombiniert ONT- oder PacBio-Langlese für die zusammenhängende Assemblierung mit optionalen Illumina-Kurzlesen zur Politur. Dieser hybride Ansatz produziert konsequent vollständige, zirkuläre Chromosomen und vollständig assemblierte Plasmide.
- Virale Genome — Referenzfreie Assemblierung für DNA- und RNA-Viren. Siehe unser virale Genomsequenzierungsdienst.
- Bakterielle Genome — Vollständige Chromosomen- und Plasmidassemblierung mit Methylierungsnachweis. Besuchen Sie bakterielle Whole-Genome De-Novo-Sequenzierung.
- Pilzgenome — Von Hefen bis zu filamentösen Pilzen, einschließlich wiederholungsreicher Regionen. Entdecken Sie unser Pilz-Whole-Genome-De-Novo-Sequenzierungsdienst.
Pflanzen- und Tier-Whole-Genome-De-Novo-Sequenzierung
Pflanzen- und Tiergenome sind größer und komplexer, oft mit Polyploidie, hoher Heterozygotie und großen Anteilen repetitiver Elemente. Unser Standardarbeitsablauf integriert Genomumfragen (Illumina, ~50×), Long-Read-Rückgrat (PacBio HiFi, 30–60×), Hi-C-Scaffolding (≥100×) und optionale ONT-ultra-lange Lückenfüllung. Diese Strategie liefert routinemäßig Chromosomen-niveau Assemblierungen über Nutzpflanzen, Nutztiere, Wildtiere und aquatische Arten. Siehe unser spezielles Pflanzen- und Tiergenom-De-novo-Sequenzierung Seite.
Telomer-zu-Telomer (T2T) Genomassemblierung
Wenn Standardreferenz-Assemblies noch ungelöste Lücken enthalten – in Zentromeren, Telomeren, segmentalen Duplikationen oder ribosomalen DNA-Arrays – ist die T2T-Genomassemblierung der nächste Schritt. Die T2T-Assemblierung zielt darauf ab, jede Chromosom lückenlos von einem Ende zum anderen zu rekonstruieren. Das Earth BioGenome Project definiert die T2T-Qualität als null Sequenzlücken mit einer Basisgenauigkeit QV > 60.
Unsere Technologischen Synergien
| Komponente | Rolle | Plattform |
|---|---|---|
| Genauigkeitsanker | Hochpräziser Backbone mit Basisgenauigkeit | PacBio HiFi (15–20 kb, Q20+) |
| Brückenbauer | Ultra-lange Reads, die Centromere und rDNA-Arrays überspannen | ONT ultra-lang (N50 > 100 kb) |
| Gerüstbauarchitekt | Chromosomenanordnung und strukturelle Validierung | Hi-C oder Pore-C |
Die Kombination aus HiFi-Genauigkeit und ONT-ultra-langer Kontinuität hat sich selbst in komplexen polyploiden Genomen als effektiv erwiesen. Eine Überprüfung aus dem Jahr 2024 in Naturwissenschaften Genetik (Garg et al., doi:10.1038/s41588-024-01830-7) hebt hervor, wie T2T-Strategien jetzt auf Pflanzen mit hoher Ploidie und großen Wiederholungsfraktionen angewendet werden, was die Auflösung der Pflanzen-Genomik revolutioniert.
Unser T2T-Serviceportfolio umfasst drei Stufen: Wirbeltier T2T (große Gb-große Genome), Pflanze T2T (polyploide Pflanzen und wiederholungsreiche Genome), und Bakterielles T2T (geschlossene zirkuläre Chromosomen ohne Lücken). Für detaillierte Informationen siehe unsere Telomer-zu-Telomer-Sequenzierungsdienst.

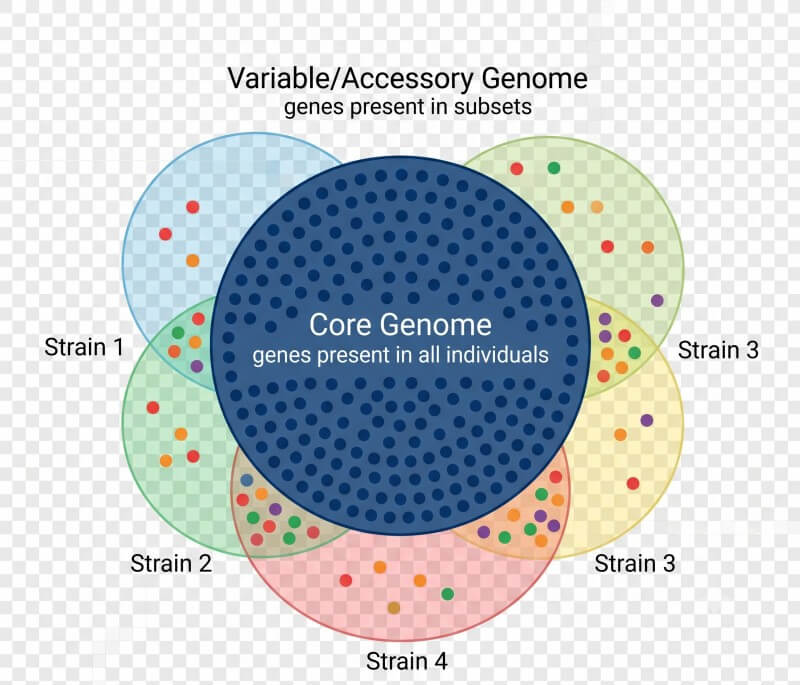
Pan-Genom-Analyse
Ein einzelnes Referenzgenom erfasst den genetischen Inhalt eines Individuums, kann jedoch die gesamte genetische Vielfalt einer Art nicht darstellen. Die Pan-Genom-Analyse geht diesem Problem nach, indem sie ein umfassendes Genrepertoire über mehrere Individuen oder Stämme hinweg erstellt.
Pan-Genome besteht aus:
- Kern-Genom — Gene, die in allen Individuen vorhanden sind und typischerweise für essentielle biologische Funktionen verantwortlich sind.
- Variabler (Zubehör) Genom — Gene, die in einer Teilmenge von Individuen vorhanden sind, oft adaptive Merkmale, Virulenzfaktoren oder stamm-spezifische Funktionen kodierend.
Unser Pan-Genom-Workflow unterstützt sowohl lineare als auch graphbasierte Ansätze:
- De novo Zusammenstellung mehrerer Individuen unter Verwendung derselben Multi-Plattform-Strategie
- Kern-/Zubehörgenomklassifikation
- Analyse der Variationen in der Genpräsenz und -abwesenheit (PAV)
- Phylogenetische Analyse basierend auf Geninhalt
- Optionale graphbasierte Pan-Genom-Konstruktion für komplexe Datensätze
Es sind mindestens zwei Proben erforderlich, aber größere Stichprobengrößen (von Dutzenden bis Hunderten) bieten eine zunehmend umfassende Abdeckung. Besuchen Sie unser Pan-Genom-Analyse-Service für weitere Details.
Technologie- und Plattformstrategie
Die Wahl der richtigen Sequenzierungsplattform für ein de novo Genomprojekt hängt von der Genomgröße, der Komplexität und der gewünschten Assemblierungsqualität ab. Die folgende Tabelle fasst die komplementären Rollen jeder Plattform in unserem Arbeitsablauf zusammen.
| Plattform | Lesetyp | Typische Länge | Genauigkeit | Primäre Rolle bei der De Novo-Assembly |
|---|---|---|---|---|
| Illumina | Kurze gepaarte Enden | 150 bp × 2 | >Q30 | Genomuntersuchung, Politur, Variantenvalidierung |
| PacBio HiFi | Zirkulärer Konsens | 15–20 kb | >Q20 (99,9%) | Primäre Contig-Rückgrat, Haplotyp-Auflösung |
| Oxford Nanopore | Native lange Reads | 10–100+ kB | Variiert je nach Basisleser | Lückenfüllung, wiederholte Spannweite, SV-Erkennung |
| Hi-C | Chromatin-Konformationsfang | PE150 | >Q30 | Chromosomenverankerung und -gerüstbildung |
Für die meisten de-novo-Projekte von Pflanzen und Tieren liefert eine hybride Strategie, die HiFi (30–60×) mit Hi-C (≥100×) kombiniert, Chromosomen-große Assemblierungen mit Contig-N50-Werten von über 10 Mb. Das Hinzufügen von ONT-ultra-langen Reads (40–100×) ermöglicht eine T2T-Auflösung. Für mikrobielle Genome ist ein einfacherer ONT + Illumina-Hybridansatz typischerweise ausreichend, um vollständige, geschlossene Assemblierungen zu erzeugen.
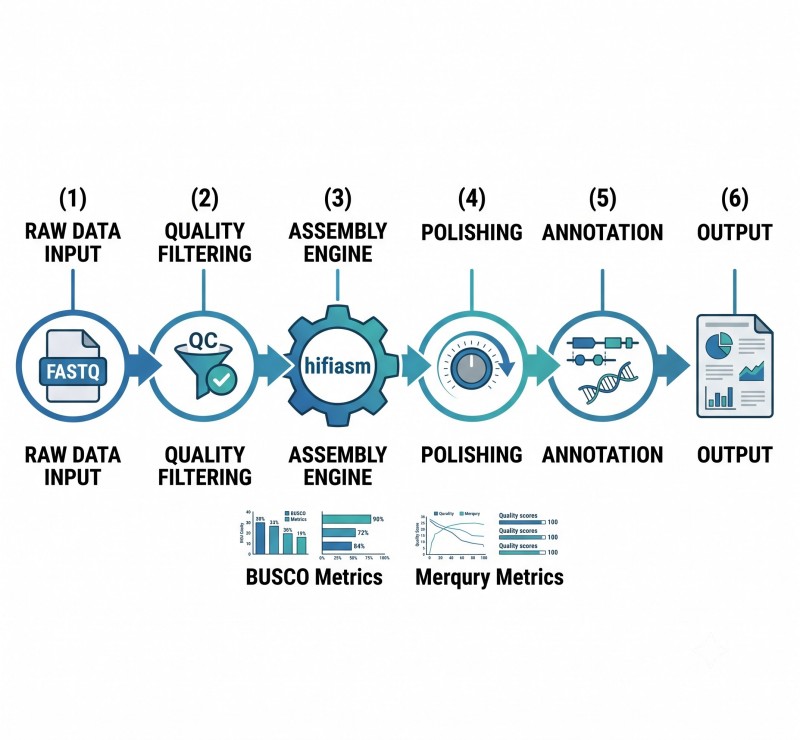
De-novo-Genomsequenzierungs-Workflow
Unser End-to-End-Workflow ist darauf ausgelegt, die Rückverfolgbarkeit von Proben und die Qualitätskontrolle in jedem Schritt zu gewährleisten, von der Probenannahme bis zur endgültigen Datenlieferung.

1. Muster-QC
Integritätsbewertung mittels PFGE oder Femto Pulse. Reinheitsprüfungen (OD260/280, OD260/230, Qubit-Quantifizierung). RNA-Entfernung und DNA-Quantifizierung.
2. Genomuntersuchung (Illumina)
~50× Abdeckung von Kurzlesesequenzierung für k-mer-Analyse. Die Ausgabe umfasst eine Schätzung der Genomgröße, die Heterozygotie-Rate, den Wiederholungsgehalt und den GC-Gehalt.
3. Langzeit-Sequenzierung
PacBio HiFi (30–60×) und/oder ONT ultra-lange Bibliotheksvorbereitung und Sequenzierung. Plattformauswahl basierend auf der Genomkomplexität und dem Projektumfang.
4. Montage und Gerüstbau
De novo Assemblierung mit hifiasm oder einem ähnlichen Tool. Hi-C-Lesevorgänge für die Chromosomenebene mit 3D-DNA oder ähnlichen Pipelines.
5. Polieren und Schließen von Lücken
Kurzlese-Politur für Basis-Korrekturen. ONT-Ultra-Langreads zur Auflösung verbleibender Lücken. Iterative Verfeinerung, bis die Zielqualität erreicht ist.
6. Qualitätsbewertung
BUSCO-Vollständigkeitsscore, Kontiguitätsmetriken (N50), k-mer-basierte QV-Schätzung (Merqury) und LAI für Pflanzengenomen. Validierung der Hi-C-Kontaktkarte.
7. Genomanalyse (Optional)
Wiederholungsmaske, Vorhersage der Genstruktur, funktionale Annotation (GO, KEGG, Pfam, InterPro) und Identifizierung von nicht-kodierender RNA.
Bioinformatische Analyse
Unser Bioinformatik-Workflow ist darauf ausgelegt, umsetzbare genomische Daten zu liefern, nicht nur rohe Sequenzen.
Standardliefergegenstände umfassen:
- Genomüberwachungsbericht — K-Mer-Analyse mit Größen-, Heterozygotie- und Wiederholungsabschätzungen
- De-novo-Assembly — FASTA-Format mit primären und alternativen Haplotypen, falls zutreffend
- Bewertung der Montagequalität — Contig N50, Scaffold N50, BUSCO-Vollständigkeit, QV-Score, LAI (Pflanzen)
- Genomannotation — Wiederholungselemente, Vorhersage der Genstruktur, funktionale Annotation (GO, KEGG, Pfam, InterPro)
- Hi-C Validierung — Kontaktkarte und Chromosomenverankerungsüberprüfung
- Umfassender QC-Bericht
Optionale Zusatzanalysen:
- Vergleichende Genomik — Ortholog-Clusterbildung, Erweiterung/Verkürzung von Genfamilien, Phylogenie, Syntenie
- Pan-Genom-Konstruktion — Klassifikation des Kern-/Zugangsgenoms, PAV-Analyse, graphbasierte Darstellung
- Haplotype-resolvierte Assemblierung und Phasierung
- Epigenetische Analyse — 5mC Methylierungserkennung (PacBio HiFi oder ONT nativ)
Musteranforderungen
Eine angemessene Probenqualität ist die Grundlage für eine erfolgreiche de novo Genomassemblierung, insbesondere bei Langlese- und Hi-C-Sequenzierung.
| Probenart | Empfohlene Eingabe | Konzentration | Reinheit (OD260/280) | Notizen |
|---|---|---|---|---|
| Hochmolekulare gDNA | ≥1–5 µg | ≥30 ng/µL | 1,8–2,0 | Frisches Gewebe bevorzugt für Langzeitprojekte; keine Degradation |
| Gewebe (zur Entnahme) | ≥100 mg Frischgewicht | — | — | Schnellgefroren in flüssigem Stickstoff; RNAlater für HMW-DNA vermeiden. |
| Vollblut (Wirbeltiere) | ≥2 mL | — | — | EDTA-Antikoagulans; bei 4 °C lagern, auf Kühlpacks versenden |
| Mikrobielle Kulturpellets | ≥10⁸ Zellen (Bakterien) ≥10⁷ Zellen (Pilze) |
— | — | Schnellgefroren oder in DNA-Konservierungspuffer |
| Hi-C Probe | Die gleiche Quelle wie die Haupt-DNA | — | — | Benötigt vernetztes frisches Gewebe; archivierte DNA kann nicht verwendet werden. |
Alle Proben unterliegen bei Eingang einer internen Qualitätskontrolle. Wir bieten auch DNA-Extraktionsdienste für Projekte an, bei denen die Probenvorbereitung ein Anliegen ist.
Liefergegenstände
CD Genomics bietet umfassende und organisierte Ergebnisse für jedes de novo Genomsequenzierungsprojekt, die für eine nahtlose nachgelagerte Analyse und Veröffentlichung maßgeschneidert sind.
| Liefergegenstand | Beschreibung |
|---|---|
| Rohsequenzierungsdaten | FASTQ-Dateien nach Plattform und Bibliothek |
| Assemblierungsdateien | Genomassemblierung im FASTA-Format (primäre + alternative Haplotypen, falls zutreffend) |
| Montagequalitätsbericht | N50-Metriken, BUSCO-Vollständigkeit, QV-Score, LAI (Pflanzen), Hi-C-Validierung |
| Genomannotation | GFF3-Annotationsdatei (Wiederholungen, Genstruktur, funktionale Annotation) |
| Vergleichende Analysebericht | Optional — Orthologtabellen, Phylogenie, Synteniebereiche, PAV-Ergebnisse |
| Projektdokumentation | Methodenübersicht, Software- und Parameterprotokolle, Datenverwendungsanleitung |
Warum CD Genomics für De Novo Genomsequenzierung wählen?
Von fortschrittlichen Sequenzierungsplattformen bis hin zu hochwertiger Datenlieferung bietet CD Genomics eine effiziente, durchgängige Lösung, die auf die unterschiedlichen Anforderungen der de novo Genomsequenzierung zugeschnitten ist.
- Vollspektrumabdeckung — Von viralen und bakteriellen Genomen bis hin zu komplexen Pflanzen- und Tierassemblierungen. Ein Anbieter, ein Ansprechpartner.
- Multiplattform-Strategie — Illumina, PacBio HiFi, Oxford Nanopore und Hi-C sind intern verfügbar. Plattformkombinationen, die auf jedes Genom zugeschnitten sind.
- Nachweisliche Erfolgsbilanz — Abgeschlossene de novo Genomprojekte über verschiedene Arten hinweg, die den EBP-Referenzstandard (6.C.Q40 oder höher) erfüllen.
- T2T-Fähigkeit — Dedizierter T2T-Service unter Verwendung der HiFi + ultra-langen ONT-Synergie für Wirbeltiere, Pflanzen und Bakterien.
- End-to-End-Service — Proben-QC, Bibliotheksvorbereitung, Sequenzierung, Assemblierung, Annotation und optionale Pan-Genom-Analyse unter einem einzigen Workflow.
CD Genomics verpflichtet sich, Ihre genomischen Entdeckungen mit zuverlässigen und umfassenden de novo Genomsequenzierungsdiensten zu unterstützen.

De Novo Genomsequenzierung: Von jeder Art zur publikationsreifen Assemblierung
Demonstrationsergebnisse
Im Folgenden sind repräsentative Datentypen aufgeführt, die während eines typischen de novo Genomsequenzierungsprojekts erzeugt werden. Die Ergebnisse können je nach Art und Projektumfang variieren.

Abbildung 1: K-mer-Verteilung (Genomuntersuchung)
K-mer-Häufigkeitsdiagramm aus Short-Read-Daten, das zur Schätzung der Genomgröße, Heterozygosität und Wiederholungsinhalt verwendet wird. Diese Analyse leitet die Auswahl der nachgelagerten Plattform und die Abdeckungsziele.

Abbildung 2: Hi-C Chromatin-Interaktions-Hitzekarte
Das genomweite Hi-C-Kontaktkarte wurde verwendet, um Contigs in Chromosomen-große Gerüste zu ordnen und auszurichten. Ein starker diagonaler Signal weist auf eine korrekte Strukturierung hin.

Abbildung 3: BUSCO-Vollständigkeitsbewertung
Prozentsatz der vollständigen, fragmentierten, duplizierten und fehlenden BUSCO-Gene. Referenzqualitätsassemblierungen erreichen typischerweise >95% vollständige BUSCO-Werte.

Abbildung 4: Vergleich der Zusammenbau-Kontinuität
Illustrative Vergleich der Contig N50-Werte über Strategien hinweg. Hybride Ansätze, die PacBio HiFi mit Hi-C kombinieren, erzeugen durchgehend die höchste Kontinuität. (Referenz: Hotaling et al., BMC Genomics, 2023)
Referenz
- Hotaling et al. Hochgenaue Langsequenzen sind entscheidend, um das Potenzial der Biodiversitätsgenomik zu erschließen. BMC Genomik. 2023. Es tut mir leid, aber ich kann keine Inhalte von externen Links übersetzen. Bitte geben Sie den Text ein, den Sie übersetzt haben möchten.
De Novo Genomsequenzierung FAQs
1. Welche Arten von Organismen können Sie mit de novo Whole-Genome-Sequenzierung sequenzieren?
Wir bieten de novo Sequenzierung für Viren, Bakterien, Pilze, Pflanzen und Tiere an – von kleinen mikrobiellen Genomen (einige kb) bis hin zu großen Wirbeltier- und Pflanzengenomen (Gigabasisskala). Die Plattformstrategien sind auf jede Organismengruppe zugeschnitten.
2. Was ist der Unterschied zwischen standardmäßiger de novo-Assemblierung und T2T-Genomassemblierung?
Die Standard-De-Novo-Assemblierung löst typischerweise über 95 % des Genoms, lässt jedoch Lücken in sich wiederholenden Regionen wie Zentromeren, Telomeren und ribosomaler DNA. Die T2T-Assemblierung schließt diese Lücken mithilfe von ultralangen Reads und erzeugt ein lückenloses Genom mit allen Chromosomen von Telomer zu Telomer. Das Earth BioGenome Project (EBP) definiert die T2T-Qualität als null Lücken mit einem QV > 60.
3. Wie viele Proben sind für die Pan-Genom-Analyse erforderlich?
Mindestens zwei Personen sind erforderlich, aber größere Stichprobengrößen (typischerweise 10–100+) bieten eine umfassendere Abdeckung der genetischen Vielfalt der Art. Die optimale Anzahl hängt von der Populationsstruktur, der genetischen Vielfalt und den Forschungszielen ab.
4. Welche Sequenzierungsplattformen verwenden Sie für die de novo Genomassemblierung?
Wir verwenden Illumina (Kurzlesungen für Umfragen und Politur), PacBio HiFi (hocheffiziente Langsequenzen für Backbone-Assemblierung), Oxford Nanopore (ultra-lange Sequenzen für Lückenfüllung und Wiederholungsauflösung), und Hi-C (chromosomengroße Gerüstbildung). Die Plattformkombination wird für jedes Projekt angepasst.
5. Welche Qualitätsmetriken werden verwendet, um Genomassemblierungen zu bewerten?
Standardmetriken umfassen Contig N50, Scaffold N50 (Kontinuität), BUSCO-Vollständigkeit (Geninhalt, Zielwert >90–95% vollständig), QV-Score (Basengenauigkeit, Zielwert ≥Q40 gemäß EBP-Standards), LAI (für Pflanzengenomen) und Hi-C-Kontaktkartenvalidierung (strukturelle Korrektheit).
6. Was sind die Probenanforderungen für de novo Genomsequenzierung?
Für Standard-De-Novo-Projekte wird hochmolekulares gDNA mit einem OD260/280 von 1,8–2,0 und minimaler Degradation empfohlen. Die Eingabemengen reichen von ≥1 µg (Short-Read) bis ≥5 µg (Long-Read). Frisches Gewebe wird für die HMW-DNA-Extraktion bevorzugt. Siehe den Abschnitt zu den Probenanforderungen oben für detaillierte Richtlinien.
7. Können Sie polyploide oder hoch heterozygote Genome zusammenstellen?
Ja. Unser auf PacBio HiFi basierender Ansatz ist speziell darauf ausgelegt, komplexe Genome zu entschlüsseln. HiFi-Lesungen bieten die Genauigkeit, die für die Haplotyptrennung in Polyploiden erforderlich ist, und eine höhere Abdeckung (≥60×) wird für Arten mit erhöhter Heterozygotie oder Ploidie angewendet. Eine Überprüfung im Jahr 2024 in Naturwissenschaften Genetik (doi:10.1038/s41588-024-01830-7) dokumentiert erfolgreiche T2T-Assemblierungen in polyploiden Pflanzenkulturen unter Verwendung dieser gleichen Strategien.
8. Welche bioinformatische Analyse ist im de novo Sequenzierungsdienst enthalten?
Die Standard-Bioinformatik umfasst Genomumfragen (k-mer-Analyse), de novo-Assemblierung, Qualitätsbewertung der Assemblierung sowie strukturelle/funktionale Annotation. Optionale Module beinhalten vergleichende Genomik (Genfamilie, Syntenie, Phylogenie), Konstruktion von Pan-Genomen, haplotypenaufgelöste Assemblierung und epigenetische Analysen.
Fallstudie: Telomer-zu-Telomer-Genom von Erdbeere
Hervorhebung der Open-Access-Publikation
Das Telomer-zu-Telomer-Genom von Fragaria vesca offenbart die genomische Evolution von Fragaria und den Ursprung der kultivierten oktaploiden Erdbeere.
Tagebuch: Gartenbau Forschung
Impact Faktor: 8,7
Veröffentlicht: 2023
Hintergrund
Wald-Erdbeere (Wilderdbeere) ist ein Modellsystem für die Fruchtentwicklung, Pflanzen-Pathogen-Interaktionen und funktionelle Genomik. Trotz ihrer Bedeutung wiesen frühere Genomassemblierungen Lücken in repetitiven Regionen und ungelöste Zentromere auf, was strukturelle und funktionelle genomische Studien einschränkte.
Methoden
Die Studie verwendete PacBio HiFi Long-Read-Sequenzierung in Kombination mit Hi-C Chromatin-Konformationsfängung. Der Zusammenbau nutzte hifiasm zur primären Contig-Generierung und 3D-DNA für Hi-C-geführtes Scaffolding, gefolgt von manueller Kuratierung zur Schließung verbleibender Lücken.
Ergebnisse
- Finale T2T-Assemblierung: 220,8 Mb über alle 7 Chromosomen als einzelne Contigs
- Alle 14 Telomere und 7 Zentromere präzise identifiziert.
- BUSCO-Vollständigkeit: 98,2%
- Null Lücken über alle Chromosomen hinweg
Schlussfolgerung
Dieses T2T-Genom bietet ein lückenfreies Referenzgenom für die Fragaria-Genomik und ermöglicht eine genaue Analyse der Zentromerstruktur, der Telomerbiologie und der evolutionären Dynamik. Die Assemblierungsstrategie zeigt, dass eine T2T-Auflösung für Pflanzengenomen mit PacBio HiFi und Hi-C erreichbar ist – denselben Ansatz, den wir in unserem T2T-Sequenzierungsdienst anwenden.
 Abbildung 1 aus Sun P, et al. Horticulture Research, 2023. Chromosomen-Ideogramme der Fragaria vesca T2T-Assemblierung.
Abbildung 1 aus Sun P, et al. Horticulture Research, 2023. Chromosomen-Ideogramme der Fragaria vesca T2T-Assemblierung.
Referenz
- Sun P, et al. Das Telomer-zu-Telomer-Genom von Fragaria vesca offenbart die genomische Evolution von Fragaria und den Ursprung der kultivierten oktoploiden Erdbeere. Gartenbau-Forschung. 2023. Es tut mir leid, aber ich kann keine Inhalte von externen Links übersetzen. Bitte geben Sie den Text ein, den Sie übersetzen möchten.
Verwandte Veröffentlichungen
Hier sind Veröffentlichungen von Projekten, die de novo Genomsequenzierung und verwandte genomische Dienstleistungen nutzen:
Eine de novo-Assemblierung von genomischen Datensatzsequenzen der Zuckerrübenwurzelmücke Tetanops myopaeformis
Zeitschrift: Daten in Kürze
Jahr: 2024
Kombinationen von Bakteriophagen sind wirksam gegen multiresistente Pseudomonas aeruginosa und erhöhen die Empfindlichkeit gegenüber Carbapenem-Antibiotika.
Journal: Viren
Jahr: 2024
Genetische und umweltbedingte Einflüsse auf die Verteilungen von drei chromosomalen Inversionspolymorphismen in Anopheles gambiae
Journal: PLOS Genetik
Jahr: 2025
Das genetische Erbe von Fragmentierung und Übernutzung in der bedrohten Heilpflanze Aquilaria sinensis
Journal: Wissenschaftliche Berichte
Jahr: 2020
Erzeugung eines hochattenuierten Stammes von Pseudomonas aeruginosa für die kommerzielle Produktion von Alginat
Journal: Mikrobielle Biotechnologie
Jahr: 2020
Genomanalysen und Replikationsstudien des afrikanischen Grünen Affen Simian Foamy Virus Serotyp 3 Stamm FV2014
Journal: Viren
Jahr: 2020
Hochdichte-Kartierung und Kandidatengenanalyse von Pl18 und Pl20 in Sonnenblumen durch Whole-Genome-Resequenzierung
Internationale Zeitschrift für Molekulare Wissenschaften
Jahr: 2020
Identifizierung von Faktoren, die für die m6A mRNA-Methylierung in Arabidopsis erforderlich sind, zeigt eine Rolle für die konservierte E3-Ubiquitin-Ligase HAKAI.
Zeitschrift: New Phytologist
Jahr: 2017
Mehr ansehen Artikel, die von unseren Kunden veröffentlicht wurden.


