
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Illumina NGS: Prinzipien, Plattformen und bewährte Verfahren für erfolgreiche Sequenzierungsprojekte
Die Illumina-Sequenzierung durch Synthese (SBS) Technologie hat über ein Jahrzehnt lang die Landschaft der Kurzzeit-Sequenzierung dominiert und treibt die Mehrheit der jährlich veröffentlichten genomischen Studien an. Die Kombination aus hoher Genauigkeit, skalierbarem Durchsatz und einem ausgereiften Ökosystem macht sie zur Standardwahl für die meisten NGS-Anwendungen – von gezielten Amplicon-Panels und Whole-Exome-Sequenzierung bis hin zu bevölkerungsbasierten Whole-Genome-Studien.
Um jedoch ein erfolgreiches Illumina-Sequenzierungsprojekt durchzuführen, ist mehr erforderlich als nur das grundlegende Workflow zu kennen. Die Auswahl der richtigen Plattform, die Vorbereitung hochwertiger Bibliotheken, die Interpretation von Qualitätsmetriken und das Vermeiden häufiger Workflow-Fehler sind entscheidend, um reproduzierbare, publikationsreife Ergebnisse zu erzielen. Eine einzige schlecht vorbereitete Bibliothek kann einen gesamten Sequenzierungslauf verschwenden – und da NovaSeq X-Flow-Zellen pro Lauf Zehntausende von Dollar kosten, ist die finanzielle Auswirkung eines Fehlers erheblich.
Dieser Artikel bietet einen praktischen Leitfaden zu Illumina NGS und behandelt die Auswahl der Plattform, bewährte Methoden zur Bibliotheksvorbereitung, die Interpretation der Sequenzierungsqualität und Arbeitsabläufe zur Datenanalyse. Er richtet sich an Forscher, die bereits die grundlegenden Prinzipien verstehen und umsetzbare Anleitungen für die experimentelle Planung und Durchführung benötigen. Der Fokus liegt durchgehend auf praktischen, entscheidungsorientierten Inhalten: welche Plattform zu wählen ist, wie man die häufigsten Fehler bei der Bibliotheksvorbereitung vermeidet, wie man einen Sequenzierungs-QC-Bericht liest und wie man ein Projekt von Anfang bis Ende plant.
Warum Illumina NGS die Landschaft der Kurzlesesequenzierung dominiert
Die Sequenzierung durch Synthese (SBS) Technologie von Illumina ist durch kontinuierliche Innovation die dominierende Plattform für Kurzlesungen geblieben. Die Chemie hat sich von der Standard-SBS zur neueren XLEAP-SBS entwickelt, die mit der NovaSeq X-Serie eingeführt wurde und schnellere Laufzeiten, höhere Signalintensität und einen reduzierten Reagenzienverbrauch bietet. Diese Verbesserungen haben die Kosten pro Genom im letzten Jahrzehnt dramatisch gesenkt und machen großangelegte Sequenzierungsprojekte wirtschaftlich machbar.
Die Illumina-Plattformfamilie umfasst einen Durchsatzbereich von 10.000-fach und deckt nahezu jede Größenordnung von Sequenzierungsprojekten ab:
| Plattform | Maximaler Ausgang | Maximale Leselänge | Typische Laufzeit | Ideale Anwendungen |
|---|---|---|---|---|
| iSeq 100 | 1,2 GB | 2 × 150 bp | 9–17,5 Std. | Kleine Paneele, Validierungsläufe |
| MiniSeq | 7,5 Gb | 2 × 150 bp | 7–24 Std. | Kleine gezielte Sequenzierung |
| MiSeq | 15 GB | 2 × 300 bp | 4–55 Std. | 16S/ITS-Amplikons, kleine Genome, Amplicon-Panels |
| NextSeq 2000 | 330 GB | 2 × 150 bp | 11–48 Std. | RNA-Seq, Exome, mittlere WGS |
| NovaSeq 6000 | 6 TB | 2 × 250 bp | 13–44 Std. | Großangelegte WGS, Bevölkerungsstudien |
| NovaSeq X / X Plus | 16 TB | 2 × 150 bp | 12–48 Std. | Ultra-große WGS, >30× menschliche Genome im großen Maßstab |
Für Forscher, die ihr erstes Illumina-Projekt planen oder ihre Plattform aufrüsten möchten, ist es entscheidend, zu verstehen, wo jedes System passt. Umfassend Next-Generation-Sequenzierungsdienste deckt die gesamte Palette der Illumina-Plattformen ab, was es ermöglicht, das richtige Instrument für die spezifischen Durchsatz- und Leseanforderungen jedes Projekts auszuwählen.
Abbildung 1: Illumina-Plattformmatrix — Durchsatz im Vergleich zur Lese-länge für MiSeq, NextSeq, NovaSeq 6000 und NovaSeq X

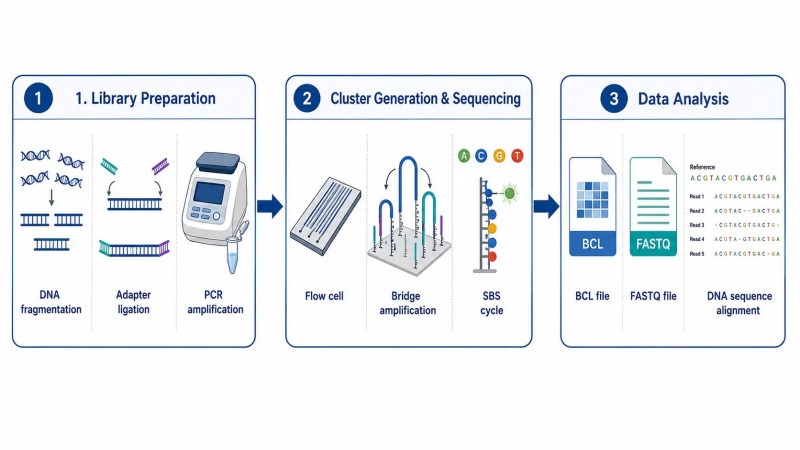
Die drei Kernschritte — Ein kurzer Überblick
Jedes Illumina-Sequenzierungsprojekt folgt dem gleichen dreistufigen Workflow:
- BibliotheksvorbereitungDNA oder RNA wird fragmentiert, endrepariert, mit A-Polymerase versehen und an Sequenzierungsadapter ligiert. Die resultierende Bibliothek wird amplifiziert, quantifiziert und auf Qualität überprüft, bevor sie geladen wird.
- Cluster-Generierung und SequenzierungBibliotheken werden auf eine Flusszelle geladen, wo sie einer Brückenamplifikation unterzogen werden, um klonale Cluster zu bilden. Die Sequenzierung durch Synthese erfolgt in Zyklen, wobei jeder Zyklus ein fluoreszenzmarkiertes, reversibel terminierendes Nukleotid einfügt. Das Instrument erfasst nach jedem Zyklus Bilder, und die Software zur Basenbestimmung wandelt die Fluoreszenzsignale in Sequenzlesungen um.
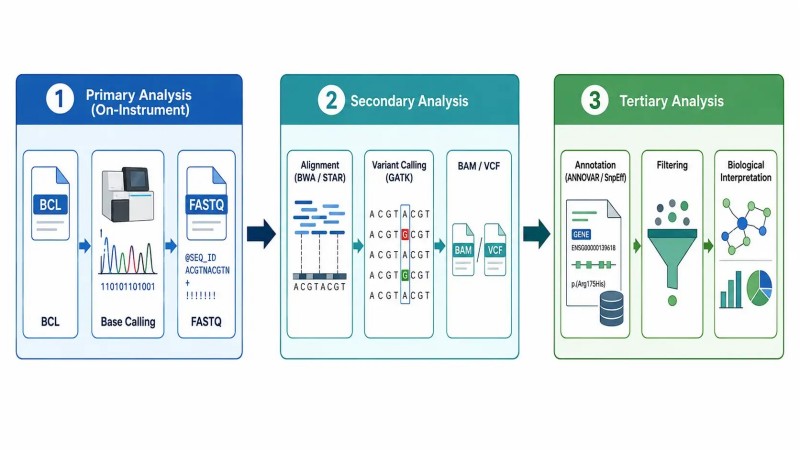
- DatenanalyseRohe BCL-Dateien werden in das FASTQ-Format konvertiert (primäre Analyse), die Reads werden an ein Referenzgenom ausgerichtet (sekundäre Analyse), und es folgt die biologische Interpretation (tertiäre Analyse).
Abbildung 2: NGS-Dreischritt-Workflow — Bibliotheksvorbereitung, Clustererzeugung und Sequenzierung
Auswahl der richtigen Illumina-Plattform — Durchsatz, Lese-länge und Anwendungsanpassung
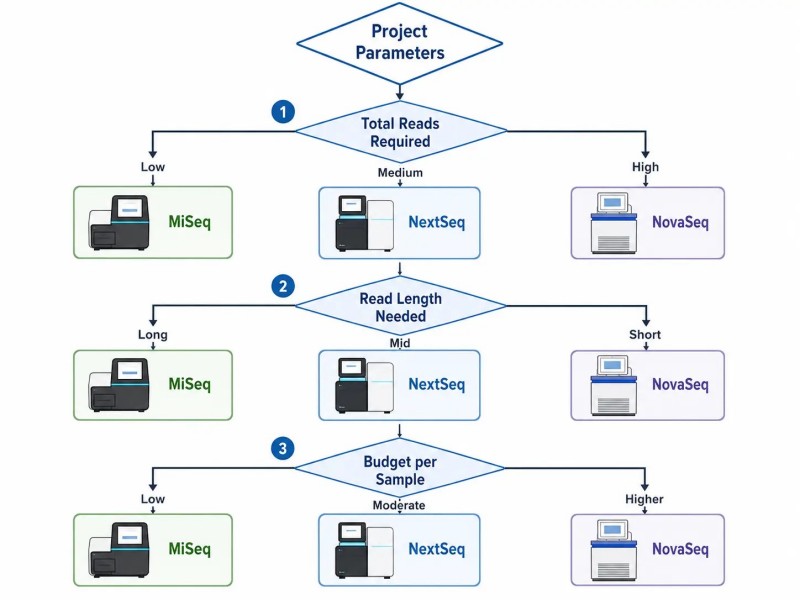
Die Wahl der falschen Plattform ist einer der häufigsten und kostspieligsten Fehler bei der Planung von NGS-Projekten. Die richtige Wahl hängt von der Interaktion zwischen drei Parametern ab: der benötigten Gesamtsequenzierungsleistung, der erforderlichen Lesegröße und dem Budget.
Anwendungsorientierte PlattformauswahlEine typische Forschungsgruppe kann Projekte über mehrere Ebenen hinweg durchführen. Das Verständnis dafür, wie jede Plattform auf gängige Studientypen abgebildet wird, gewährleistet eine effiziente Ressourcennutzung.
- 16S/ITS Amplicon-SequenzierungErfordert 2 × 250 bp oder 2 × 300 bp Reads, um die vollständigen variablen Regionen abzudecken. MiSeq ist die Standardplattform, die 96–384 Proben pro Durchgang zu geringen Kosten pro Probe verarbeitet.
- Whole-Exom-Sequenzierung (WES)Benötigt ~10 Gb pro Probe. Für 96 Proben verarbeitet eine NovaSeq 6000 S4 Flusszelle die gesamte Charge in einem Durchlauf. Für kleinere Chargen von 12–24 Proben ist der NextSeq 2000 praktischer und vermeidet die Zahlung für ungenutzte Flusszellenkapazität.
- Whole-Genome-Sequenzierung (WGS)Benötigt 30–60 Gb pro Probe für 30× Abdeckung. NovaSeq 6000 oder NovaSeq X sind die geeigneten Plattformen. Der NovaSeq X mit XLEAP-SBS-Chemie hat die Kosten für die Sequenzierung pro Genom erheblich gesenkt, wodurch ultra-große WGS-Studien zugänglicher geworden sind.
- RNA-Seq (mRNA)Erfordert 20–50 Millionen Reads pro Probe für die standardmäßige Genexpression; 100+ Millionen für die Analyse auf Isoformebene. Die NextSeq 2000 eignet sich gut für Standardprojekte, während Plattformen der NovaSeq-Klasse Einzelzell-RNA-seq-Projekte unterstützen, die 500 Millionen bis 3 Milliarden Reads pro Durchlauf benötigen.
- Gezielte Panels (klein)10–100 Gene mit 1–5 Millionen Reads pro Probe. MiniSeq oder MiSeq sind kosteneffektiv und bieten eine schnelle Bearbeitungszeit. Für Panels mit mehr als 500 Amplicons könnte NextSeq erforderlich sein, um eine ausreichende Lesetiefe pro Amplicon sicherzustellen.
Praktischer EntscheidungsrahmenBeginnen Sie mit der Berechnung der Gesamtzahl der benötigten Reads (Reads pro Probe × Anzahl der Proben). Überprüfen Sie dann die minimale Read-Länge. Wählen Sie schließlich die Plattform, die diesen Durchsatz in der kürzesten Zeit zu den niedrigsten Kosten pro Probe liefert. NGS-Sequenzierungsdienste Das Team kann Ihnen helfen, Ihre Projektparameter mit der optimalen Plattformkonfiguration abzugleichen.
Abbildung 3: Entscheidungsbaum zur Plattformauswahl — von Projektparametern zum empfohlenen Illumina-System
Bibliotheksvorbereitung — Der Schritt, an dem die meisten Projekte erfolgreich sind oder scheitern
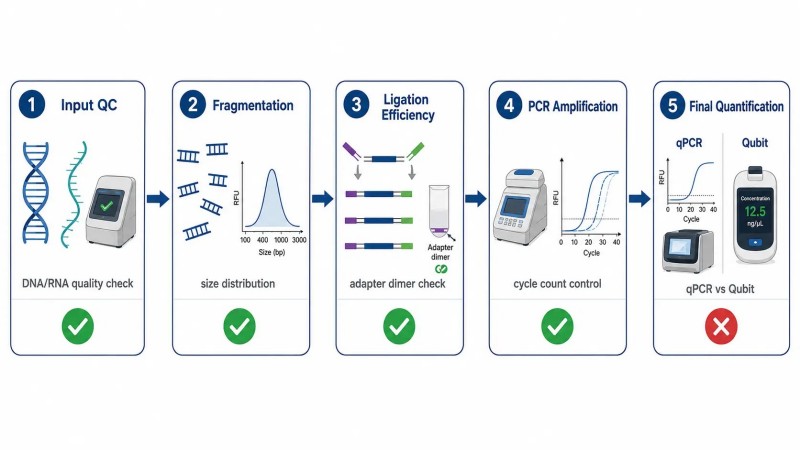
Die Bibliotheksvorbereitung ist der variabelste Schritt im NGS-Workflow und die häufigste Quelle für Projektfehler. Ein gut gestaltetes Protokoll zur Bibliotheksvorbereitung mit strengen QC-Prüfpunkten ist entscheidend für konsistente Ergebnisse.
Fünf kritische QC-Prüfpunkte:
- Eingangs-NukleinsäurequalitätDNA sollte ein OD260/280 von 1,8–2,0 und ein OD260/230 > 1,5 haben. RNA sollte einen RIN ≥ 7 für mRNA-seq und einen RIN ≥ 5 für total RNA-seq aufweisen. Degradiertes Ausgangsmaterial ist die häufigste Ursache für das Scheitern von Bibliotheken und kann nicht durch Erhöhung der Ausgangsmenge kompensiert werden.
- FragmentierungskonsistenzDie enzymatische Fragmentierung ist für die meisten Anwendungen reproduzierbarer als mechanisches Scheren. Die Zielverteilung der Fragmentgrößen sollte mit der Sequenzierleselänge übereinstimmen – für 2 × 150 bp sollte die Einfügungsgröße um 300–500 bp zentriert sein.
- Adapter-LigationseffizienzIneffiziente Ligation erzeugt Bibliotheken mit hohem Adapter-Dimer-Gehalt. Ein Bioanalyzer-Diagramm, das einen ausgeprägten Peak bei 80–120 bp zeigt, ohne dass ein entsprechendes Insert vorhanden ist, weist auf Adapter-Dimere hin, die die Sequenzierungskapazität verschwenden und die Datenqualität verringern.
- PCR-AmplifikationsbiasBegrenzen Sie die PCR-Zyklen auf 6–10 für DNA-Bibliotheken und 12–15 für RNA-Bibliotheken. Übermäßige Amplifikation erhöht die Duplikationsraten, ohne die Komplexität der Bibliothek zu verbessern. Für Proben mit niedrigem Input sollten Sie PCR-freie Bibliotheksvorbereitungsverfahren in Betracht ziehen.
- Endgültige BibliotheksquantifizierungDie qPCR-basierte Quantifizierung ist genauer als Qubit oder Bioanalyzer zur Bestimmung der Lade-Konzentration. Eine Diskrepanz von 2–3× zwischen den Methoden ist häufig, und sich auf die falsche Messung zu verlassen, ist eine der Hauptursachen für eine schlechte Clusterdichte.
Häufige Bibliotheksfehler und deren Lösungen:
- Niedrige Cluster-DichteDie Bibliothekskonzentration wurde unterschätzt. Validieren Sie die Quantifizierung mit qPCR. Für gemusterte Flusszellen (NovaSeq) ist der optimale Ladebereich eng – eine Abweichung von 10–20 % kann zu schlechten Ergebnissen führen.
- ÜberclusteringDie Bibliothekskonzentration wurde überschätzt. Neu quantifizieren und bei einer niedrigeren Konzentration neu zusammenfassen. Übermäßiges Clustern führt zu überlappenden Clustern, die nicht aufgelöst werden können, was die Anzahl der verwendbaren Reads verringert.
- Adapter-Dimer-Kontamination in ReadsDie Nachreinigung nach der Ligation war unzureichend. Erhöhen Sie das Verhältnis der SPRI-Perlen oder fügen Sie einen gelbasierten Größenauswahl-Schritt hinzu. Bei hartnäckigen Fällen verwenden Sie eine doppelseitige SPRI-Reinigung. Ein Bioanalyzer-Diagramm mit einem dominanten Peak unterhalb des erwarteten Bibliotheksgrößenbereichs bestätigt eine Kontamination mit Adapter-Dimeren.
- Hohe Duplikationsrate (>30%)Unzureichendes Eingangs-DNA oder zu viele PCR-Zyklen. Erhöhen Sie das Eingangsmaterial, falls verfügbar; reduzieren Sie die PCR-Zyklen; oder wechseln Sie zu einem PCR-freien Bibliotheksprotokoll für WGS-Anwendungen.
- Index-HoppingAuf gemusterten Flusszellen können verbleibende freie Indizes benachbarte Cluster falsch annotieren. Verwenden Sie einzigartige doppelte Indizes (UDI) anstelle von Einzelindizes, um das Problem des Index-Hoppings auszuschließen. Für große multiplexierte Projekte mit vielen Proben wird UDI dringend gegenüber Einzelindex-Strategien empfohlen.
Multiplexing-Strategie und Barcode-ZuweisungEine entscheidende Entscheidung bei der Bibliotheksvorbereitung ist, wie viele Proben pro Sequenzierlauf multiplexiert werden sollen. Die Anzahl der Proben pro Lauf wird durch die erforderlichen Reads pro Probe und die Gesamtausgabe der Flusszelle bestimmt. Bei einem NextSeq 2000, der 400 Millionen Reads generiert, ist es unkompliziert, 96 Exomproben mit jeweils 4 Millionen Reads zu multiplexieren. Bei einem MiSeq, der 25 Millionen Reads generiert, kann das Multiplexen von mehr als 48 Proben für ein 16S-Amplicon-Projekt zu unzureichenden Reads pro Probe führen, um zuverlässige Diversitätsschätzungen zu erhalten.
Die Qualität der Indizes ist ein weiterer oft übersehener Faktor. Niedrigqualitative Indizes mit hoher Ähnlichkeit zwischen den Barcode-Sequenzen erhöhen das Risiko von Fehlzuweisungen. Die Verwendung validierter Indizesätze vom Hersteller der Bibliotheksvorbereitung – mit einem minimalen Hamming-Abstand von 3 zwischen beliebigen zwei Indizes – minimiert die Übertragung zwischen Proben im selben Lauf.
Für Teams, die die Bibliotheksvorbereitung auslagern möchten, genomische Datenanalyse-Dienste Bibliothek QC und Vorbereitung als Teil eines umfassenden Sequenzierungs-Workflows einbeziehen.
Abbildung 4: QC-Workflow zur Bibliotheksvorbereitung — fünf kritische Qualitätskontrollpunkte vom Eingangs-DNA bis zur finalen Bibliotheksquantifizierung
Verständnis des Sequenzierens durch Synthese (SBS) Zyklus im Detail
Während die Einführung das grundlegende SBS-Prinzip abdeckte, ist das Verständnis der zyklusbezogenen Mechanik nützlich für die Fehlersuche und die Interpretation von QC-Metriken.
Jeder SBS-Zyklus durchläuft vier Schritte: (1) Incorporation — ein fluoreszenzmarkierter, reversibel terminierter Nukleotid wird von der Polymerase hinzugefügt; (2) Imaging — das Instrument bildet die Oberfläche der Flusszelle bei vier Wellenlängen ab, um zu identifizieren, welcher Basen an jedem Cluster eingebaut wurde; (3) Cleavage — der fluoreszierende Farbstoff und die terminierende Gruppe werden entfernt; (4) Wash — nicht incorporierte Reagenzien werden vor dem nächsten Zyklus ausgespült.
Die benötigte Zeit pro Zyklus variiert je nach Plattform. Auf dem NovaSeq 6000 dauert jeder Zyklus etwa 5–10 Minuten, einschließlich der Bildaufnahmezeit. Auf dem NovaSeq X mit XLEAP-SBS-Chemie wird die Zykluszeit auf 3–5 Minuten verkürzt, dank schnellerer Enzymkinetik und eines neu gestalteten Bildgebungssystems, das die gesamte Flusszelle in weniger Aufnahmen erfasst.
Der Hauptfehlermodus auf Zyklusebene ist "Phasierung" und "Vor-Phasierung". Phasierung tritt auf, wenn einige Vorlagen in einem Cluster es nicht schaffen, ein Nukleotid in einem bestimmten Zyklus einzufügen, und um eine Base zurückbleiben. Vor-Phasierung tritt auf, wenn einige Vorlagen in einem einzigen Zyklus zwei Basen einfügen und dadurch vorausziehen. Beide Effekte verringern die Synchronität des Clusters und führen zu einem Signalverfall über aufeinanderfolgende Zyklen. Dies ist der grundlegende Grund, warum die Qualitätswerte gegen Ende eines Reads abnehmen – nicht aufgrund eines Instrumentenfehlers, sondern als natürliche Folge unvollkommener Synchronität in einem chemischen Prozess mit mehreren Zyklen.
Phasierungsraten werden typischerweise als Prozentsatz pro Zyklus ausgedrückt. Eine Phasierungsrate von 0,1 % bedeutet, dass nach 100 Zyklen 10 % der Vorlagen in jedem Cluster um eine Base hinter der Mehrheit liegen. Nach Zyklus 150 wächst dies auf 15 %. Der kumulative Effekt bestimmt die praktische Leselängenbegrenzung für jede Plattform. Die höchste Phasierungsspezifikation von Illumina liegt typischerweise bei <0,5 % pro Zyklus für die Standard-SBS-Chemie und ist niedriger für XLEAP-SBS.
Illumina-Plattformen verwalten das Phasing durch proprietäre Algorithmen, die den Prozentsatz der Moleküle schätzen und korrigieren, die voraus oder hinterher laufen. Wenn jedoch die Zyklenanzahl über 150–300 Zyklen (je nach Plattform) steigt, verringert sich der kumulierte Effekt sowohl auf die Q-Scores als auch auf die nutzbare Leseweite. Aus diesem Grund kann NovaSeq X mit XLEAP-SBS, das reduzierte Phasing-Raten aufgrund schnellerer Kinetik und verbesserter Waschprozesse aufweist, höhere Q-Scores über längere Reads aufrechterhalten im Vergleich zur Standard-SBS-Chemie.
Verständnis der Sequenzierungsqualität — Q-Werte, Fehlerprofile und Daten-QC
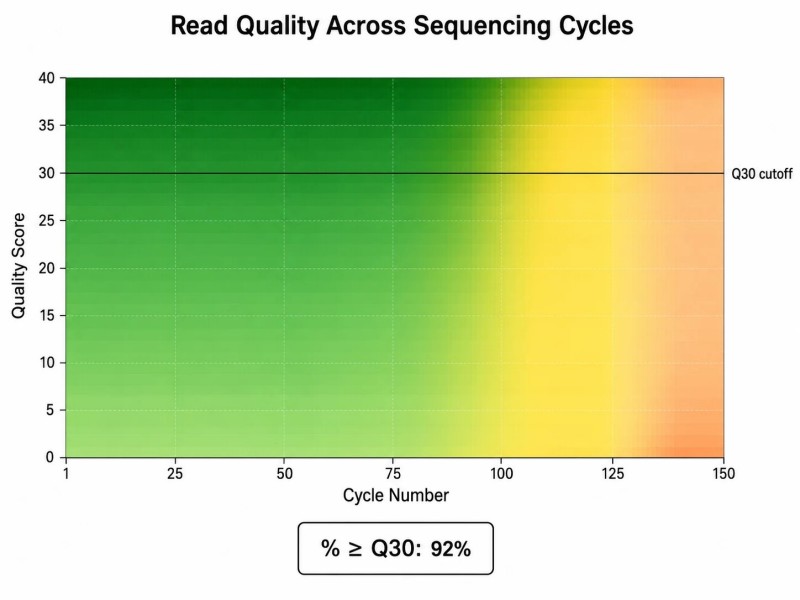
Qualitätsbewertungen (Q-Scores) Das primäre Maß zur Bewertung der Leistung eines Illumina-Sequenzlaufs ist der Phred-Qualitätswert (Q), der logarithmisch mit der Wahrscheinlichkeit eines fehlerhaften Basenaufrufs verknüpft ist: Q30 entspricht einer Fehlerwahrscheinlichkeit von 1/1000 (99,9% Genauigkeit), während Q20 einer Fehlerwahrscheinlichkeit von 1/100 (99% Genauigkeit) entspricht. Der Wert wird berechnet als Q = -10 log₁₀(P), wobei P die Wahrscheinlichkeit eines fehlerhaften Basenaufrufs ist.
Für einen typischen Illumina-Lauf zeigen die folgenden Benchmarks eine gute Leistung an:
- >85 % der Basen bei Q30 oder höher für 2 × 150 bp Läufe
- >75 % der Basen bei Q30 für 2 × 250 bp oder längere Läufe
- Fehlerquote (PhiX-Ausrichtungsabweichung) < 1%
Interpretation eines Sequenzierungs-QC-BerichtsDer Standard-Illumina-Analyse-Viewer bietet mehrere wichtige Kennzahlen, die nach jedem Lauf überprüft werden sollten:
- Qualitäts-Hitzekarte pro ZyklusZeigt die Q-Score-Verteilung über alle Zyklen. Ein allmählicher Rückgang von Anfang bis Ende ist normal; ein plötzlicher Abfall in der Mitte des Laufs kann auf ein Reagenzien- oder Fluidikproblem hinweisen.
- Basenzusammensetzung nach ZyklusFür ausgewogene Bibliotheken sollten sich die A- und T-Kurven überlappen, ebenso wie die G- und C-Kurven. Eine Divergenz weist auf eine Bias in der Bibliothekszusammensetzung hin, insbesondere bei Amplicon- oder Anreicherungs-Panels.
- GC-GehaltverteilungEin unimodaler Peak, der mit dem erwarteten GC-Gehalt des Zielgenoms übereinstimmt, weist auf eine normale Bibliothekskomplexität hin. Mehrere Peaks oder eine breite flache Verteilung deuten auf Kontamination oder PCR-Bias hin.
- DuplikationsrateFür WGS-Bibliotheken liegen die erwarteten Duplikationsraten bei 5–15 %. Höhere Raten deuten auf eine geringe Eingangs-DNA, übermäßige PCR oder unzureichende Bibliothekskomplexität hin.
Faktoren, die die Qualitätsbewertungen beeinflussenMehrere Parameter während des Sequenzierungslaufs beeinflussen die endgültige Q-Score-Verteilung. Das Verständnis dieser Faktoren hilft sowohl bei der Planung von Experimenten als auch bei der Fehlersuche bei schlechten Läufen.
- LesepositionDie Qualität nimmt gegen Ende der Lesung ab, da sich der Zerfall des fluoreszierenden Signals ansammelt und die Phaseneffekte ausgeprägter werden. Die letzten 5–10 Zyklen eines 150 bp-Lesens zeigen typischerweise niedrigere Q-Werte als die ersten 50 Zyklen. Dies ist normal und zu erwarten – die Abnahmerate ist ein nützliches Diagnosemittel.
- SequenzzusammensetzungGC-reiche Regionen und Homopolymer-Trakte neigen aufgrund der verringerten Nukleotidvielfalt während der Bildgebung zu einer niedrigeren Qualität. Das Hinzufügen von PhiX-Kontrolle (5–20 % der gesamten Bibliotheksmasse) zu Bibliotheken mit geringer Diversität bietet ein ausgewogenes Signalreferenz, das die Qualitätswerte während des gesamten Laufs erheblich verbessert.
- ClusterdichteSowohl Unter- als auch Überclusterung verringern die Qualität. Der optimale Dichtebereich variiert je nach Plattform – für NovaSeq 6000 S4 Flusszellen sind 250–350 K Cluster/mm² typisch. Für NextSeq 2000 sind 150–250 K Cluster/mm² optimal. Eine Abweichung von mehr als 20 % vom optimalen Bereich führt typischerweise zu einem messbaren Rückgang der Q30-Prozentsätze.
- IndexsequenzvielfaltNiedrig-Diversitätsindex-Sequenzen (z. B. ausschließlich A oder ausschließlich T) können während der ersten Sequenzierungszyklen des Indexlesens zu Registrierungsfehlern führen. Die Verwendung eines vorab gestalteten, validierten Indexsatzes vom Hersteller des Bibliotheksvorbereitungs-Kits vermeidet dieses Problem vollständig.
- Reagenqualität und LagerungAbgelaufene oder unsachgemäß gelagerte Sequenzierungsreagenzien sind eine häufige, verborgene Ursache für Qualitätsminderung. SBS-Chemie ist empfindlich gegenüber Gefrier-Tau-Zyklen und Temperaturschwankungen. Die Befolgung der Lager- und Handhabungsrichtlinien des Herstellers – sowie das Protokollieren der Reagenzien-Lotnummern und Verfallsdaten – ist ein einfacher, aber oft übersehener Schritt.
Die Überprüfung des Sequenzierungs-QC-Berichts, bevor mit der Datenanalyse fortgefahren wird, ist entscheidend. Wichtige Abschnitte umfassen die Qualitäts-Hitzekarte pro Zyklus, die Basenzusammensetzung nach Zyklus, die GC-Gehaltverteilung und die Duplikationsrate. Wenn eine Kennzahl außerhalb der akzeptablen Bereiche liegt, sollte der Lauf markiert und die Ursache untersucht werden, bevor die Daten für die nachgelagerte Analyse verwendet werden.
Abbildung 5: Typische Illumina Q-Score-Wärmekarte, die die Qualitätsverteilung pro Zyklus über einen 2 × 150 bp Lauf zeigt.
NovaSeq X und XLEAP-SBS-Chemie – Was sich geändert hat und warum es wichtig ist
Die Einführung der NovaSeq X-Serie mit der XLEAP-SBS-Chemie im Jahr 2023 stellt das bedeutendste Update der Illumina-Chemie seit einem Jahrzehnt dar. XLEAP-SBS ist keine geringfügige Überarbeitung – es handelt sich um eine neu gestaltete Sequenzierungschemie mit messbaren Verbesserungen in Bezug auf Geschwindigkeit, Genauigkeit und Kosten. Der NovaSeq X Plus kann bei voller Kapazität bis zu 16 Tb Daten pro Lauf generieren, was der Sequenzierung von mehr als 500 menschlichen Genomen mit 30× Abdeckung in einem einzigen 48-Stunden-Lauf entspricht.
Wesentliche Verbesserungen gegenüber dem Standard-SBS:
- Schnellere EnzymkinetikXLEAP-SBS-Enzyme integrieren Nukleotide schneller, wodurch die Laufzeiten von 2 × 150 bp von etwa 40 Stunden (NovaSeq 6000) auf etwa 24 Stunden (NovaSeq X) reduziert werden.
- Verbesserte SignalintensitätEin höheres Signal-Rausch-Verhältnis reduziert die Fehlerraten, insbesondere in den späteren Zyklen von langen Reads. Veröffentlichten Daten von Illumina zufolge zeigen sich im Vergleich zu standardmäßigem SBS auf dem NovaSeq 6000 eine Reduktion der Fehlerraten um 30-40 %.
- Reduzierter ReagenzienverbrauchDie neue Chemie verwendet weniger Reagenz pro Basis, was die Kosten pro Gb im Vergleich zur Standard-SBS-Chemie erheblich senkt.
- Höhere Durchsatzrate pro DurchlaufDie 25B- und 100B-Flow-Zellen unterstützen zuvor unmögliche Skalierungen – ein einzelner NovaSeq X Plus-Lauf kann 16 Tb Daten erzeugen, was etwa 500 menschlichen Genomen bei 30× Abdeckung entspricht.
Praktische Implikationen für ForscherDie NovaSeq X ersetzt nicht alle vorherigen Illumina-Plattformen. Für kleinere Projekte (weniger als 50 Proben) bleiben MiSeq und NextSeq aufgrund ihrer niedrigeren Mindestlaufkosten und schnelleren Bearbeitungszeiten praktischer. Die NovaSeq X ist transformativ für Projekte, die großangelegte, kosteneffiziente Sequenzierung erfordern – Bevölkerungsstudien, longitudinale Kohortenanalysen und Einzelzell-Atlas-Projekte.
Abbildung 6: XLEAP-SBS gegenüber standardisierter SBS-Chemie — wesentliche Verbesserungen in Geschwindigkeit, Signalintensität und Reagenzienverbrauch

NGS-Datenanalyse — Von BCL zu biologischen Erkenntnissen
Die Datenanalyse-Pipeline für Illumina-Sequenzierung folgt einer standardmäßigen dreistufigen Struktur:
Primäranalyse (instrumentengestützt)Das Sequenziergerät führt eine Echtzeit-Basenanrufung durch, indem es Fluoreszenzbilder in BCL (Binary Base Call)-Dateien umwandelt und diese dann in das FASTQ-Format konvertiert. Dieser Schritt ist vollständig automatisiert und erfordert typischerweise keine Benutzerintervention. Moderne Plattformen bieten Echtzeit-Qualitätsmetriken, die während des Laufs zugänglich sind.
Sekundäranalyse (benutzerverwaltet)FASTQ-Dateien werden durch Ausrichtung (STAR für RNA, BWA-MEM für DNA, HISAT2 für Transkriptome) und Variantenaufruf (GATK, FreeBayes, Strelka2) verarbeitet. Diese Phase erfordert 32–64 GB RAM für menschliches WGS und erheblichen Speicherplatz – ein einzelnes 30× menschliches Genom erzeugt etwa 100–200 GB FASTQ-Daten und etwa 50–100 GB ausgerichtete BAM-Dateien.
Tertiäre Analyse (biologische Interpretation)Annotierte Varianten werden gefiltert, priorisiert und im biologischen Kontext der Studie interpretiert. Zu den gängigen Werkzeugen für die tertiäre Analyse gehören ANNOVAR, SnpEff und VEP zur Annotation sowie eine Vielzahl von Paketen für die Analyse von Wegen und Anreicherungen.
Kritische Überlegungen zur Datenanalyse:
- ReferenzgenomversionGRCh38 (mit Patches) bleibt der Standard für das menschliche Referenzgenom. Die T2T-CHR13-Referenz bietet eine vollständigere Darstellung, ist jedoch noch nicht universell anerkannt. Die Ergebnisse der Pipeline können zwischen den Referenzversionen erheblich variieren.
- LagerplanungEin typisches WGS-Projekt benötigt 3–5× den Roh-FASTQ-Speicher für Zwischenablagen. Planen Sie 600 GB–1 TB pro 30× menschlichem Genom ein, einschließlich FASTQ, BAM, VCF und temporären Pipeline-Dateien.
- RecheninfrastrukturCloud-basierte Analysen (AWS, Google Cloud oder dedizierte Bioinformatik-Plattformen) werden zunehmend gegenüber lokalen Servern für große Projekte bevorzugt, wodurch die Notwendigkeit einer Kapitalinvestition in Computerhardware entfällt. Der Hauptnachteil ist die Datenübertragungszeit – das Hochladen von 10 TB FASTQ-Dateien kann je nach Verbindungsgeschwindigkeit 2 bis 5 Tage in Anspruch nehmen. Hybride Ansätze (lokaler Speicher + Cloud-Computing) sind bei großangelegten Projekten üblich.
- Pipeline-ReproduzierbarkeitDie Verwendung von containerisierten Pipelines (Docker, Singularity) oder Workflow-Managern (Nextflow, Snakemake, Cromwell) stellt sicher, dass dieselbe Analyse konsistent auf alle Proben in einem Projekt angewendet wird. Dies ist entscheidend für die Aufrechterhaltung der Datenvergleichbarkeit, insbesondere in Multi-Batch- oder kollaborativen Studien.
Für Forschungsteams ohne interne Bioinformatik-Kapazitäten, Genomdatenanalyse-Dienste Zugang zu etablierten Pipelines für Alignment, Variantenaufruf und biologische Interpretation bereitstellen.
Abbildung 7: Drei-Schichten-NGS-Datenanalyse-Pipeline — von BCL über FASTQ zu ausgerichtetem BAM bis zur biologischen Interpretation
Planung eines erfolgreichen Illumina-Sequenzierungsprojekts – Ein schrittweises Rahmenwerk
Über die technischen Details jedes Arbeitsablaufs hinaus teilen erfolgreiche Illumina-Projekte einen gemeinsamen Planungsrahmen. Die Einhaltung dieser Struktur minimiert das Risiko kostspieliger Änderungen oder Wiederholungen während des Projekts.
- Definieren Sie die biologische Frage und bestimmen Sie den optimalen Assaytyp. Handelt es sich um eine Entdeckungsstudie (WGS, RNA-seq), eine gezielte Nachverfolgung (WES, gezielte Panel) oder eine Screening-Anwendung (Amplicon-Panel)? Der Assay-Typ bestimmt alle nachgelagerten Parameter.
- Berechne die erforderliche Sequenzierungstiefe. Für die menschliche WGS sind 30× für die meisten Keimbahn-Anwendungen ausreichend. Die Erkennung seltener Varianten kann 60× erfordern. RNA-seq zur Genexpressionsanalyse benötigt 20–50 Millionen Reads pro Probe; die Analyse auf Isoform-Ebene erfordert über 100 Millionen. Zielgerichtete Panels benötigen 500–1.000× Abdeckung pro Amplicon für eine zuverlässige Variantenbestimmung.
- Wählen Sie die Plattform und die Flusszelle aus. Passen Sie die gesamte Leseanforderung (Reads pro Probe × Anzahl der Proben + 10–20 % Übersequenzierung) an die verfügbaren Plattformen an. Die ausgewählte Plattform sollte den erforderlichen Durchsatz liefern, ohne ungenutzte Kapazität. Ein MiSeq-Lauf, der 15 Gb erzeugt, ist für kleine Amplicon-Studien geeignet, aber ineffizient für ein großes Exomprojekt; eine NovaSeq X-Flusszelle mit Terabase-Skalenausgabe ist überdimensioniert für eine kleine Pilotstudie.
- Entwerfen Sie Bibliotheken mit QC-Prüfpunkten. Plan für Bioanalyzer-Spuren nach der Fragmentierung und nach der endgültigen Bibliothek, qPCR-Quantifizierung und einem kleinen Pilotversuch zur Titration neuer Bibliothekstypen. Jeder Kontrollpunkt sollte ein vordefiniertes Bestehen/Nichtbestehen-Kriterium haben.
- Einschließlich experimenteller Kontrollen. Eine positive Kontrollprobe mit bekannten Varianten validiert den Workflow von der Bibliotheksvorbereitung bis zur Variantenbestimmung. Eine negative (ohne Vorlage) Kontrolle identifiziert Kontamination. PhiX-Spike-in (typischerweise 1 % für WGS, 5–20 % für niedrigdiverse Bibliotheken wie Amplicons) bietet einen Kalibrierungsstandard für die Qualitätsbewertung.
- Planen Sie die Datenanalyse, bevor die Sequenzierung beginnt. Die Auswahl der Pipeline, die Version des Referenzgenoms, die Rechenressourcen und die Speicherkapazität sollten alle vorhanden sein, bevor der erste Sequenzierungslauf abgeschlossen ist. Die Sequenzierung erzeugt Daten schneller, als die meisten Forscher erwarten – ein NovaSeq X, der in 48 Stunden 16 Tb produziert, erfordert entsprechend schnelle nachgelagerte Kapazitäten.
Häufige Workflow-Fehler und wie man sie vermeidet
| Fehlermodus | Ursache | Prävention |
|---|---|---|
| Niedrige Cluster-Dichte | Bibliothekskonzentration unterschätzt; qPCR-Quantifizierung ungenau | Verwenden Sie qPCR für die endgültige Quantifizierung; führen Sie einen Titrationstest für neue Bibliothekstypen durch; validieren Sie dies mit Qubit. |
| Über-Klusterung | Bibliothekskonzentration überschätzt; gemusterte Flusszellenbeladung zu hoch | Validieren Sie mit zwei orthogonalen Methoden; konservativ verdünnen; beginnen Sie in der Mitte des empfohlenen Ladebereichs. |
| >30% Duplikationsrate | Unzureichende Eingangs-DNA; zu viele PCR-Zyklen; geringe Bibliothekskomplexität | Verwenden Sie nach Möglichkeit ≥100 ng Eingangs-DNA; begrenzen Sie die PCR-Zyklen auf ≤8; ziehen Sie eine PCR-freie Bibliotheksvorbereitung für WGS in Betracht. |
| Index-Hopping | Rückstandsfreie Adapterindizes auf gemusterten Flusszellen | Verwenden Sie einzigartige doppelte Indizes (UDI) anstelle von einzelnen Indizes; UDI beseitigt das Risiko des Index-Hüpfens vollständig. |
| Niedriges Q30 in den letzten Zyklen | Die Lese-Länge überschreitet den effektiven chemischen Bereich; Phasenakkumulation | Verwenden Sie die empfohlene maximale Leselänge der Plattform; führen Sie einen Pilotversuch durch, bevor Sie die vollständige Produktion starten. |
| Adapterkontamination in Reads | Unvollständige Reinigung nach der Adapterligatur; kurze Insertfragmente | Optimieren Sie das Verhältnis der SPRI-Perlen; fügen Sie eine gelbasierte Größenwahl für problematische Probenarten hinzu. |
| PhiX-Abgleichsrate >2% | Reagenzabbau; Flusszellenfehler; Drift bei der Basenaufrufkalibrierung | Protokollieren Sie die Chargennummern und Verfallsdaten der Reagenzien; überprüfen Sie die Flusszelle; kalibrieren Sie neu, wenn das Problem weiterhin besteht. |
Jeder Fehlermodus hat eine spezifische Grundursache und eine klare präventive Maßnahme. Probleme frühzeitig zu erkennen durch Kleinserienversuche – das Testen von Ladekonzentrationen über 3–4 Verdünnungen vor der Vollproduktion – verhindert die teuersten Sequenzierungsfehler.
Wie CD Genomics Illumina NGS-Projekte unterstützt
CD Genomics bietet umfassende Illumina-Sequenzierungsdienste an, die den gesamten Projektablauf von der experimentellen Planung bis zur Datenübermittlung abdecken.
PlattformverfügbarkeitUnser Labor ist mit NovaSeq X Plus, NovaSeq 6000, NextSeq 2000 und MiSeq-Systemen ausgestattet, die den gesamten Durchsatzbereich von kleinen gezielten Panels bis hin zu bevölkerungsweiten WGS abdecken. Jede Plattform wird unter strengen QC-Protokollen gewartet, um eine konsistente Datenqualität zu gewährleisten. Unsere Plattformwahl wird von Ihren Projektparametern bestimmt – nicht von dem, was wir zur Verfügung haben, denn wir haben jedes Illumina-System in aktiver Nutzung.
Umfassende BibliotheksvorbereitungWir bieten Standard-, Low-Input-, PCR-freie und Ultra-Low-Input-Bibliotheksvorbereitungsprotokolle an, die für verschiedene Probenarten optimiert sind – einschließlich Blut, Gewebe, FFPE, cfDNA und Einzelzellen. Qualitätskontrollen in jeder Phase.
Datenanalyse und -interpretationStandardlieferungen umfassen FASTQ-Dateien mit QC-Berichten sowie optionale sekundäre Analysen (BWA/GATK-Pipeline, RNA-seq-Quantifizierung) und tertiäre Analysen (Variantennotation, funktionelle Anreicherung). Für größere Projekte können wir cloudbasierte Analyse-Pipelines bereitstellen, die sich an Ihr Datenvolumen anpassen.
ProjektberatungUnser Team hilft dabei, die Parameter Ihres Projekts mit der optimalen Plattform, der Flow-Zellen-Konfiguration und der Sequenzierungsstrategie abzugleichen, um die Datenqualität zu maximieren und die Kosten zu minimieren. Eine typische Beratung umfasst: erwartete Datenmenge, optimale Lese- und Abdeckungsstärke, Laufkonfiguration (einzelne vs. gepaarte Enden), Multiplexing-Strategie und Anforderungen an die Datenanalyse.
Für weitere Details erkunden Sie unser NGS-Dienstleistungen oder kontaktieren Sie unser Team für eine projektbezogene Beratung.
Häufig gestellte Fragen (FAQ)
Was ist der Unterschied zwischen SBS- und XLEAP-SBS-Chemie?
XLEAP-SBS ist eine neu gestaltete Sequenzierungschemie, die mit NovaSeq X eingeführt wurde. Sie bietet schnellere Laufzeiten, höhere Signalintensität und einen geringeren Reagenzienverbrauch im Vergleich zur standardmäßigen SBS-Chemie, die auf früheren Illumina-Plattformen verwendet wurde.
Wie wähle ich zwischen MiSeq, NextSeq und NovaSeq für mein Projekt?
Beginnen Sie mit der Berechnung Ihres gesamten Lesebedarfs (Reads pro Probe × Anzahl der Proben). MiSeq eignet sich für kleine Panels und Amplicon-Projekte. NextSeq passt zu mittelgroßen Projekten wie RNA-seq und Exom-Sequenzierung. NovaSeq-Klassenplattformen sind für großangelegte WGS- und Bevölkerungsstudien konzipiert.
Welche Cluster-Dichte sollte ich auf einem NovaSeq 6000 S4-Flusszelle anstreben?
Der optimale Bereich liegt typischerweise bei 250–350 K Clustern pro mm². Werte außerhalb dieses Bereichs können den Datenertrag oder die Qualität verringern.
Warum ist mein Sequenzierungs-Q30-Score niedriger als erwartet?
Häufige Ursachen sind: Bibliothek mit geringer Nukleotidvielfalt (mehr PhiX hinzufügen), Über- oder Unterclusterung, degradierte Eingangs-DNA/RNA oder die Verwendung einer Lesegröße, die den optimalen Bereich der Plattform überschreitet.
Wie kann ich feststellen, ob meine Bibliotheksvorbereitung vor der Sequenzierung erfolgreich war?
Eine erfolgreiche Bibliothek sollte einen klaren Peak im Bioanalyzer-Trace im erwarteten Größenbereich zeigen, minimale Kontamination mit Adapterdimeren (<5% der Bibliotheksmasse) aufweisen und konsistente qPCR-Quantifizierungsergebnisse liefern.
Was verursacht Adapter-Dimere und wie entferne ich sie?
Adapter-Dimere bilden sich, wenn Adaptermoleküle sich aneinander binden, anstatt an die Insert-DNA. Sie können entfernt werden, indem das Verhältnis der SPRI-Perlen während der Reinigung erhöht wird oder indem ein gelbasiertes Größenauswahlverfahren hinzugefügt wird.
Was ist der Unterschied zwischen Index-Hopping und Index-Crosstalk?
Index-Hopping tritt auf, wenn verbleibende freie Index-Primer benachbarte Cluster falsch annotieren, wodurch Reads von einer Probe in einer anderen erscheinen. Index-Crosstalk resultiert aus Signalinterferenzen zwischen Indexsequenzen während der Bildgebung. UDI (einzigartige doppelte Indizes) beseitigt Index-Hopping effektiv.
Welche Datenausgabe sollte ich von einem menschlichen WGS 30× Lauf erwarten?
Ungefähr 90–100 Gb Rohdaten pro Probe, was ~100–200 GB FASTQ-Dateien, ~50–100 GB ausgerichtete BAM-Dateien und ~1–2 GB gVCF-Dateien ergibt.
Wie viel Speicherplatz benötige ich für ein NGS-Projekt?
Planen Sie für das 3–5-fache der Roh-FASTQ-Größe, um Zwischenanalyse-Dateien unterzubringen. Für ein WGS-Projekt mit 100 Proben bedeutet dies insgesamt 30–50 TB Speicherplatz.
Welche Referenzgenomversion sollte ich für menschliche Sequenzierungsdaten verwenden?
GRCh38 ist der aktuelle Standard für die meisten Anwendungen. T2T-CHR13 ist vollständiger, wird jedoch noch nicht von allen Analysetools unterstützt. Passen Sie die Referenzversion an die Anforderungen der Tools und die Standards der Gemeinschaft für Ihre spezifische Anwendung an.
Referenzen
- Übersicht über den Illumina NGS-Workflow. Illumina, Inc.
- Qualitätsbewertungen für Next-Generation-Sequenzierung. Illumina Technisches Dokument.
- Sequenzierungsfehlerprofile von Illumina-Sequenziergeräten. NAR Genomik und Bioinformatik. 2021;3(1):lqab019.
- Chemie und Bildgebung auf der NovaSeq X-Serie. Illumina Wissensdatenbank.
- Cluster-Dichte-Optimierung auf Illumina-Sequenzierinstrumenten. GenoHub.
Verwandte Dienstleistungen
- Nächste Generation Sequenzierung
- Genomdatenanalyse
- Whole Genome Sequenzierung
- Genotypisierung
- Bioinformatik-Dienstleistungen
- RNA-Seq
- Mikrobielle Sequenzierungsdienstleistung


