
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Präzise Verknüpfungskartierung: Hochdichte Markerintegration und Rekombinationsanalyse in komplexen Genomen
Die Hochdichte-Verknüpfungskartierung wird oft als ein Problem der Markeranordnung beschrieben. In der Praxis handelt es sich jedoch um ein Problem der Rekombinationsinferenz. Die eigentliche Herausforderung besteht nicht nur darin, mehr SNPs entlang eines Chromosoms zu platzieren, sondern auch zu entscheiden, wie viel echte meiotische Information diese SNPs enthalten. Diese Unterscheidung wird entscheidend in großen, repetitiven oder polyploiden Genomen, wo die Markerhäufigkeit viel schneller zunehmen kann als die Rekombinationsauflösung.
Diese Ressource behandelt die Linkage-Mapping-Workflows für die genomische Analyse zu Forschungszwecken und die nachgelagerte biologische Interpretation. Sie beschreibt nicht die klinische Diagnose, die Patienteneinstufung oder die therapeutische Entscheidungsfindung.
Eine moderne Verknüpfungskarte ist eine statistische Rekonstruktion der Chromosomenübertragung während der Meiose. Jedes Intervall wird aus den Segregationsmustern abgeleitet, die in einer Kartierungspopulation beobachtet werden. Jede abgeleitete Distanz hängt von Annahmen über den Abstand der Crossing-over, die Genotyp-Sicherheit, fehlende Daten, Allel-Dosierung, lokale Marker-Redundanz und das Vererbungsmodell der Art ab. Wenn diese Annahmen schwach sind, kann eine dichte Karte präzise erscheinen, während sie biologisch instabil bleibt.
Die wichtigste Unterscheidung in diesem Bereich ist nicht niedrige Dichte versus hohe Dichte. Es ist Marker-Dichte versus RekombinationsdichteEin Chromosom kann Zehntausende von aufrufbaren Varianten enthalten, jedoch nur eine begrenzte Anzahl informativer Crossing-Over in den verfügbaren Nachkommen. Wenn die Analyse jeden Marker als unabhängiges Positionshinweis behandelt, wird die lokale Auflösung überbewertet. Wenn stattdessen das Chromosom als eine Reihe von durch Rekombination unterstützten Erbblöcken modelliert wird, wird die Karte viel zuverlässiger.
Dieses Problem ist in einfachen diploiden Systemen leicht zu unterschätzen. Bei moderater Genomgröße, moderater Wiederholungsbelastung und relativ sauberer Segregation können herkömmliche Arbeitsabläufe weiterhin gut funktionieren. Aber sobald das Projekt in Arten mit großen Genomen, strukturell ungleichen Chromosomen oder polyploider Vererbung übergeht, beginnen ältere Annahmen zu versagen. Crossing-over wird sichtbar ungleichmäßig. Interferenz spielt eine Rolle. Markersysteme beproben das Genom nicht gleichmäßig. Genotypisierung mit geringer Tiefe kann Allel-Kopienzustände verwischen. An diesem Punkt hört die Linkage-Kartierung auf, eine einfache Genotypisierungsübung zu sein, und wird zu einer Übung in biologischer Zurückhaltung.
Eine nützliche Möglichkeit, über die Verknüpfungsanalyse nachzudenken, besteht darin, drei Ebenen aufeinander abzustimmen. Die erste Ebene ist die meiotische Biologie: homologe Paarung, Synapsis, Crossover-Zuweisung, Interferenz und Chromatin-Kontext. Die zweite Ebene ist die Messung: wie Marker generiert werden, wo sie fallen, wie viel Lese-Tiefe sie unterstützt und wie oft technisches Rauschen Rekombination imitiert. Die dritte Ebene ist die Inferenz: Markeranordnung, Wahl der Kartenfunktion, Haplotyp-Phasierung, Bin-Konstruktion und QTL-Modellierung. Die meisten schlechten Karten entstehen nicht durch zu wenig Daten. Sie entstehen durch Fehlanpassungen zwischen diesen Ebenen.
Das ist auch der Grund, warum die Verknüpfungsanalyse selten allein in einem ernsthaften Genomik-Workflow steht. Sobald stabile Intervalle festgelegt sind, fließt die Karte oft direkt in umfassendere Variantenentdeckungs- und Genominterpretationspipelines ein. In Projekten, die eine dichte Entdeckung von genomweiten Polymorphismen vor der Kartenkonstruktion benötigen, Whole-Genome-Sequenzierung kann ein breites Varianten-Substrat bereitstellen, während Variantenerkennung wird entscheidend für die Umwandlung von Rohsequenzdaten in ein Markerset, das für eine erbschaftsbasierte Analyse geeignet ist. Die Karte ist wertvoll, nicht weil sie viele Marker enthält, sondern weil sie eine vertrauenswürdige nachgelagerte Interpretation unterstützt.
Warum die Rekombinationsbiologie die Karte verankern muss
Jede Verknüpfungskarte liegt downstream von der Meiose. Das klingt offensichtlich, dennoch verhalten sich viele Pipelines immer noch so, als wäre Rekombination nur eine statistische Störung, die die Software später beheben kann. Das ist sie nicht. Die Software kann nur die Crossing-Over-Historien interpretieren, die die Population tatsächlich erzeugt hat. Wenn diese Historien spärlich, strukturiert oder stark eingeschränkt sind, wird eine höhere Marker-Dichte allein keine zusätzlichen Informationen liefern.
Die Rekombination beginnt mit programmierten Doppelstrangbrüchen während der meiotischen Prophase I. Diese Brüche werden verarbeitet und repariert, aber nur einige reifen zu Crossover heran. Eine Verknüpfungskarte erfasst nicht direkt jedes Brechereignis. Sie erfasst die Erbfolgen der Crossover-Ergebnisse, die durch die Gametenbildung überleben und in Nachkommen gemessen werden können. Dies ist wichtig, da eine niedrige beobachtete Rekombination nicht immer dasselbe bedeutet. Eine Region kann genetisch komprimiert erscheinen, weil Brüche selten sind, weil die Reparatur ohne Crossover dominiert, weil die lokale Chromatinstruktur einschränkend ist, weil die Chromosomenstruktur den Austausch unterdrückt oder weil ein nahegelegenes Crossover bereits die Wahrscheinlichkeit eines weiteren Ereignisses durch Interferenz verringert hat.
Diese Unterscheidung ist nicht akademisch. Sie bestimmt, ob ein markerreicher Intervall tatsächlich informativ ist. Wenn eine Region physisch mit SNPs gesättigt ist, aber biologisch arm an Nachweis von Crossing-over, kann die scheinbare Detailgenauigkeit der Karte irreführend werden. Der wahre Engpass ist nicht die Sequenzierungskapazität. Es ist die meiotische Gelegenheit.
Chiasmata, Chromosomenarchitektur und die Grenzen der lokalen Auflösung
Die strukturelle Logik der Prophase I erklärt, warum homologe Chromosomen entlang proteinreicher Achsen ausgerichtet sind und durch den Synaptonemal-Komplex verbunden werden. Diese Architektur stabilisiert die Paarung und bietet den räumlichen Kontext, in dem die Crossing-over-Stellen festgelegt werden. Das spätere Chiasma ist die zytologische Spur dieses früheren molekularen Prozesses.
Aus kartografischer Sicht bedeutet dies, dass Crossover-Positionen keine freien Variablen sind. Sie werden durch die Chromosomenorganisation geprägt. Lange Chromosomen erfordern in der Regel mindestens ein Crossover für eine ordnungsgemäße Segregation, dennoch neigen Crossover-Ereignisse dazu, sich nicht eng zu gruppieren. Dies ist ein Grund, warum dichte Karten häufig auf eine harte Obergrenze bei der lokalen Verfeinerung stoßen. Eine Region kann viele Marker enthalten, aber wenn die verfügbaren Meiosen nur wenige distincte Bruchpunkte dort erzeugt haben, kann die Analyse keine echte Trennung über das hinaus erzwingen, was die Biologie bereitgestellt hat.
Die praktische Konsequenz ist wichtig. Viele instabile lokale Anordnungen in dichten Karten sind keine Anzeichen dafür, dass das Chromosom ungewöhnlich kompliziert ist. Sie sind Anzeichen dafür, dass die Analyse von einer Region eine positionsgenaue Präzision verlangt, die nie genügend informative Rekombination erzeugt hat. In solchen Fällen besteht die richtige Reaktion oft darin, die Region auf der Ebene von ko-segregierenden oder nahe ko-segregierenden Einheiten zusammenzufassen, anstatt darauf zu bestehen, dass jeder benachbarte SNP eine eindeutig aufgelöste Position hat.
Mapping-Funktionen sind verborgene Annahmen über den Abstand zwischen den Crossover-Punkten.
Die Umwandlung von Rekombinationsfraktion in Kartenentfernung sieht zwar nach einem technischen Detail aus, ist aber tatsächlich ein kompaktes Modell dafür, wie Crossing-Over entlang des Chromosoms verteilt ist.
Die Haldane-Funktion geht davon aus, dass Kreuzungen unabhängig auftreten. Nach diesem Modell beeinflusst eine Kreuzung nicht die Wahrscheinlichkeit einer anderen nahegelegenen Kreuzung. Mehrere verborgene Kreuzungsereignisse werden im Rahmen eines Zufallsereignismodells behandelt. Historisch gesehen war dies elegant und nützlich. Aber es beschreibt ein Chromosom ohne Interferenz.
Die Kosambi-Funktion geht davon aus, dass die Platzierung von Crossover nicht vollständig zufällig ist. Sie berücksichtigt einen Grad an Interferenz, was bedeutet, dass ein Crossover die Wahrscheinlichkeit eines anderen in der Nähe verringert. In vielen biologischen Systemen führt dies zu Abständen, die plausibler sind als die, die aus einem strikten Modell ohne Interferenz abgeleitet werden.
Dennoch sollte keine der Funktionen als automatische Wahrheit betrachtet werden. In Karten mit niedriger Dichte mag der praktische Unterschied gering erscheinen. In Karten mit hoher Dichte akkumuliert sich der wiederholte lokale Bias. Die Gesamtlänge der Karte ändert sich. Die Intervallskala ändert sich. QTL-Spitzen können breiter oder schmaler erscheinen, als es die zugrunde liegende Rekombinationsstruktur rechtfertigt. Eine früh in der Kartenkonstruktion getroffene Modellwahl kann daher die scheinbare Präzision jeder nachfolgenden Schlussfolgerung beeinflussen.
Die richtige Gewohnheit besteht darin, Mapping-Funktionen als konkurrierende biologische Hypothesen zu betrachten. Wenn eine Art starke Crossover-Interferenz, distale Rekombinationskonzentration, geschlechtsspezifische Rekombinationsunterschiede oder chromosomenklassenspezifisches Verhalten zeigt, sollte der Workflow die Sensitivität gegenüber diesen Annahmen testen, anstatt eine Softwarevorgabe ohne Überprüfung zu übernehmen. Die beste Funktion ist nicht die, die die ordentlichsten Ausgaben produziert. Es ist diejenige, die eine stabile Ordnung, eine kohärente Intervallstruktur und eine verteidigbare biologische Interpretation liefert.
Rekombinationsinterferenz ist ein Entwurfsprinzip, kein Korrekturterm.
Interferenz wird oft eingeführt, um zu erklären, warum beobachtete doppelte Crossing-over seltener sind als erwartet. Diese Definition ist für moderne dichte Karten zu eng. Interferenz wird besser als ein leitendes Prinzip des Abstands zwischen Crossing-overs verstanden.
In praktischen Begriffen führt Interferenz dazu, dass Crossover gleichmäßiger verteilt sind, als es ein zufälliges Modell vorhersagen würde. Sobald ein Crossover festgelegt ist, wird es für nahegelegene Stellen weniger wahrscheinlich, ein weiteres zu beherbergen. Dies beeinflusst, wie viele verschiedene rekombinante Klassen in einer Mapping-Population erscheinen und damit, wie viel lokale Ordnungsinformation ein Chromosom bereitstellen kann.
Deshalb bleiben einige markerreiche Intervalle genetisch komprimiert, selbst wenn die Sequenzierungstiefe ausreichend und die Markerbestimmung technisch einwandfrei ist. Das Fehlen lokaler Bruchpunkte könnte die Biologie der Interferenz widerspiegeln, anstatt einen Fehler des Tests darzustellen. Ohne diese Perspektive könnten Forscher niedrige Bruchpunktsegmente fälschlicherweise als schlechte Datenregionen interpretieren und weiterhin Marker zu einem Problem hinzufügen, das grundlegend biologischer Natur ist.
Interferenz verändert auch, wie die Auflösung beurteilt werden sollte. Die Auflösung steigt nicht linear mit der Anzahl der Marker. Sie nimmt mit der Anzahl und Platzierung informativer Überlappungsgeschichten zu. Sobald Interferenz die Bildung nahegelegener Überlappungen einschränkt, beginnen dichte Marker-Panels oft, Redundanz statt neuer Informationen zu messen.
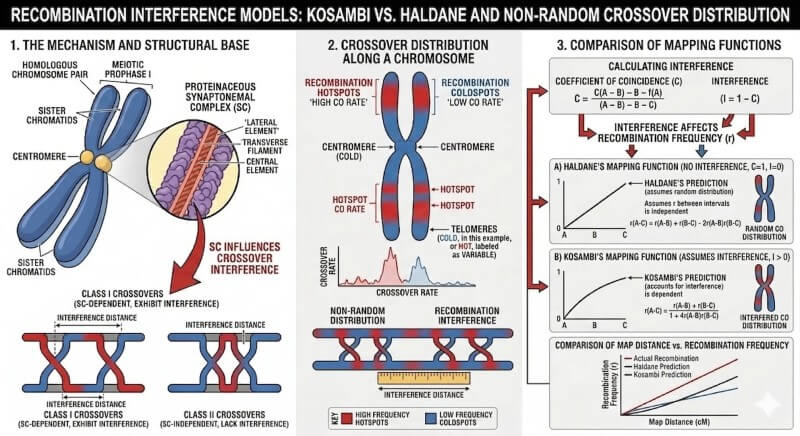 Abbildung 1. Interferenz sorgt dafür, dass der Abstand zwischen den Crossover-Ereignissen regelmäßiger ist, als es ein zufälliges Modell vorhersagt, sodass markerreiche Regionen genetisch komprimiert bleiben können, wenn die Meiose nur wenige distincte lokale Bruchpunkte hervorbringt. Die Abbildung verdeutlicht, warum dichte Genotypisierung nicht automatisch eine feinkörnige Kartenauflösung erzeugt.
Abbildung 1. Interferenz sorgt dafür, dass der Abstand zwischen den Crossover-Ereignissen regelmäßiger ist, als es ein zufälliges Modell vorhersagt, sodass markerreiche Regionen genetisch komprimiert bleiben können, wenn die Meiose nur wenige distincte lokale Bruchpunkte hervorbringt. Die Abbildung verdeutlicht, warum dichte Genotypisierung nicht automatisch eine feinkörnige Kartenauflösung erzeugt.
Die praktische Implikation ist einfach: Wenn dichte Marker keine zusätzlichen rekombinanten Strukturen mehr offenbaren, sollte die Karte diesen Grenzwert widerspiegeln. Dies ist der Punkt, an dem die Co-Segregationslogik und später die binbasierte Abstraktion analytisch notwendig und nicht mehr optional werden.
Hotspots, Coldspots und die ungleiche Geometrie des Chromosoms
Ein Chromosom ist keine einheitliche Rekombinationsoberfläche. Einige Segmente fungieren als Hotspots, an denen Überkreuzungen mit erhöhter Frequenz auftreten. Andere verhalten sich als Coldspots, bei denen lange physische Abschnitte nur wenig zur genetischen Distanz beitragen.
Das ist wichtig, weil eine Kopplungskarte den Rekombinationsraum misst, nicht den physischen Raum. Zwei Intervalle, die in Megabasen ähnlich aussehen, können in Zentimorgans radikal unterschiedlich erscheinen, wenn eines in einer hotspot-reichen Region liegt und das andere in einer Rekombinationswüste. Infolgedessen garantiert selbst eine physisch gleichmäßige Platzierung der Marker keine gleichmäßige genetische Auflösung.
Die Zugänglichkeit der Chromatinstruktur ist ein Hauptgrund dafür. Offenes Chromatin ist im Allgemeinen permissiver gegenüber der meiotischen Maschinerie, die die Rekombination einleitet und verarbeitet. Wiederholungsreiche oder heterochromatische Segmente sind oft weniger permissiv. In einigen Wirbeltier-Systemen helfen PRDM9-Bindungsmotive dabei, die Positionen von Hotspots zu bestimmen. In vielen Pflanzengenomen ist die Architektur der Hotspots enger mit der Zugänglichkeit in der Nähe von Promotoren und dem lokalen Sequenzkontext verbunden. Die genauen Determinanten unterscheiden sich zwischen den Taxa, aber die kartografischen Konsequenzen sind konsistent: Das Chromosom ist genetisch heterogen.
Diese Heterogenität erklärt, warum einige Marker-Plattformen je nach Studienziel stärker erscheinen als andere. Eine Strategie mit reduzierter Repräsentation kann bevorzugt zugängliche Sequenzen erfassen und daher Marker in Regionen anreichern, die bereits häufiger rekombinieren. Das kann hilfreich sein, wenn das Hauptziel eine effiziente QTL-Erkennung in genreichen Intervallen ist. Es kann jedoch auch einen irreführenden Eindruck von ausgewogener genomweiter Abdeckung erzeugen.
Die nützlichere Frage ist nicht, ob Marker das Chromosom physisch überspannen. Es ist vielmehr, ob sie die Rekombinationsmöglichkeiten erfassen, wo die Studie Auflösung benötigt. Diese Unterscheidung wird entscheidend, wenn es darum geht, ein Markersystem für große und komplexe Genome auszuwählen.
Auswahl von Markersystemen für große und komplexe Genome
Die häufigste Plattformfrage heute ist, ob man Genotypisierung durch Sequenzierung oder Low-Pass-Ganzgenom-Skim-Sequenzierung verwenden sollte. Locker formuliert klingt das nach einer Wahl zwischen kostengünstiger reduzierter Repräsentation und breiterer Genomabdeckung. Für die Kopplungsanalyse ist die Entscheidung spezifischer als das. Die eigentliche Frage ist, welche Plattform die am besten interpretierbaren Rekombinationsevidenzen für die Art, das Populationsdesign und die nachgelagerten Ziele liefert.
Bei Genomen, die größer als 10 Gb sind, wird dies zu einer strategischen Entscheidung anstelle einer technischen Präferenz. Sehr große Genome verdünnen die Lesetiefe über einen massiven physischen Raum. Wiederholungsinhalte erschweren die Ausrichtung. Kopie-variable oder niedrigkomplexe Regionen können die Genotyp-Sicherheit destabilisieren. Unter diesen Bedingungen beeinflusst die Wahl der Plattform nicht nur die Anzahl der Marker, sondern auch die Fehlerrate, das lokale Vertrauen, die Dosierungsinferenz und die Arten von Schlussfolgerungen, die die Karte später unterstützen kann.
Genotypisierung durch Sequenzierung: wenn gezielte Komplexitätsreduktion ein Vorteil ist
GBS reduziert die Genomkomplexität vor der Sequenzierung. Durch die Fokussierung auf eine Teilmenge von restriktionsdefinierten Fragmenten konzentriert es die Reads in einem handhabbaren Darstellungsraum. Für große biparentale Populationen schafft dies oft ein günstiges Kosten-zu-Informations-Verhältnis. Eine Kopplungskarte benötigt keine umfassende Sequenzabdeckung. Sie benötigt informative segregierende Loci über viele Individuen hinweg.
Deshalb Genotypisierung durch Sequenzierung (GBS) häufig gut abschneidet, wenn das unmittelbare Ziel die Erstpass-Kartenkonstruktion in einer großen Population ist und das Budget durch die Stichprobengröße und nicht durch die Notwendigkeit einer genomweiten physischen Kontinuität eingeschränkt ist. Wenn Hunderte von Nachkommen typisiert werden müssen, kann die Fähigkeit, die Kosten pro Probe niedrig zu halten und gleichzeitig eine nützliche Tiefe an ausgewählten Loci aufrechtzuerhalten, unvollständige physische Abdeckung überwiegen.
Aber GBS hat sichtbare Grenzen. Die Marker-Wiederherstellung hängt von der Verteilung der Restriktionsstellen und dem Verhalten der Bibliothek ab. Fehlende Daten sind oft strukturiert, nicht zufällig. Loci können in genreichen oder zugänglichen genomischen Regionen gruppiert sein, während wiederholungsreiche oder rekombinationsarme Bereiche unterproben werden. In diploiden Projekten können diese Verzerrungen tolerierbar sein. In komplexen Genomen können sie zu interpretativen Verzerrungen werden.
Niedrigdurchlass-Ganzgenom-Skim-Sequenzierung: wenn der breite physische Kontext wichtiger ist
Low-Pass-Whole-Genome-Skim-Sequenzierung erfasst das gesamte Genom mit einer geringen durchschnittlichen Tiefe. Ihre Stärke liegt in der Breite. Sie kann Marker über einen größeren physischen Anteil des Genoms bereitstellen und ist oft wiederverwendbarer für das Verankern von Gerüsten, die Bewertung des strukturellen Kontexts und spätere haplotypbasierte Analysen.
Das macht Skim-Sequenzierung attraktiv, wenn erwartet wird, dass die Karte mehrere Rollen erfüllt. Wenn das Projekt später möglicherweise die Unterstützung von Montagevalidierung, langfristiger Intervallinterpretation oder umfassender genomweiter Haplotyprekonstruktion benötigt, kann Skim-Daten einen Wert bieten, der über die ursprüngliche Karte hinausgeht.
Die Schwäche ist ebenso klar. In sehr großen Genomen kann die Skim-Tiefe so dünn werden, dass Heterozygoten unterbewertet, Dosierungszustände verschwommen und falsche Rekombinationen nach aggressivem Hard-Calling oder Imputation eingeführt werden. Eine breite physische Abdeckung ist nur hilfreich, wenn die Unsicherheit der Genotypen ehrlich modelliert wird. Wenn eine niedrige Tiefe so behandelt wird, als wäre sie eine saubere diskrete Genotypisierung, kann die Karte breiter, geräuschhafter und weniger vertrauenswürdig werden als ein gezielteres Datenset.
Entscheidungskriterien: Wann jede Plattform wahrscheinlich helfen oder scheitern wird.
Der nützlichste Weg, um zwischen GBS und Skim-Sequenzierung zu wählen, besteht darin, den Engpass der Studie klar zu definieren.
Wenn das Projekt ist budgetbeschränkt und stichprobenreichGBS hat oft den Vorteil. Es konzentriert die Lesevorgänge, unterstützt größere Nachkommenschaftsätze und kann genügend Marker für eine effektive Verknüpfungsrekonstruktion wiederherstellen, ohne für eine vollständige Genomdarstellung zu bezahlen.
Wenn das Projekt es erfordert späteres Gerüstverankern, physische Intervallwiederverwendung oder umfassendere Haplotypinterpretation, wird das Skim-Sequenzieren trotz der rauschhaften Rohdaten attraktiver. Seine physische Breite kann die zusätzliche Komplexität rechtfertigen, wenn die Karte nur eine Komponente eines größeren genomischen Workflows ist. In assemblierungsorientierten Studien kann diese Logik auch mit Hi-C-Sequenzierung, insbesondere wenn eine langfristige chromosomale Struktur über die Kopplungskarte hinaus erforderlich ist.
Wenn das Projekt beinhaltet polyploide Vererbung oder starke Abhängigkeit von dosissensitiver GenotypisierungDie Wahl wird vorsichtiger. Flache Skim-Daten können fehlschlagen, wenn Allel-Kopienzustände nicht zuverlässig getrennt werden können. In diesem Szenario kompensiert der breite physische Fußabdruck des Skim-Sequenzierens nicht für instabile Genotypnachweise. Ebenso kann GBS fehlschlagen, wenn Locus-Ausfälle, strukturierte Fehlstellen oder eingeschränkte Repräsentation zu wenig Unterstützung für homolog-spezifische Phaseninferenz bieten.
Eine einfache Regel hilft. Wählen Sie die Plattform, die am besten die die zerbrechlichste Variable In Ihrem Design. Wenn die fragile Variable die Probenanzahl ist, gewinnt GBS oft. Wenn es um die Intervallwiederverwendung in nachgelagerten genomischen Aufgaben geht, könnte das Skim-Sequencing gewinnen. Wenn es um die Genotyp-Sicherheit in einem dosis-sensitiven System geht, sollte zuerst die Plattform ausgeschlossen werden, die keine zuverlässige Allel-Zustands-Inferenz aufrechterhalten kann.
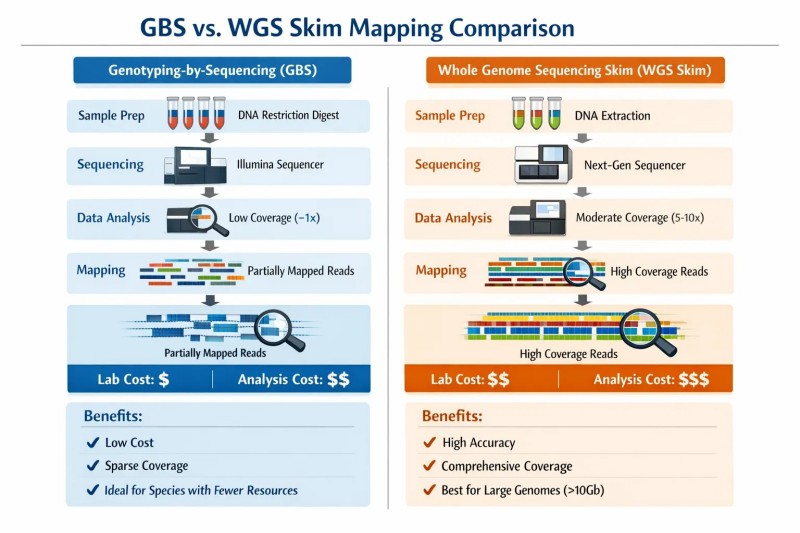 Abbildung 2. Der eigentliche Kompromiss besteht nicht darin, "günstig versus umfassend", sondern in der Genotyp-Sicherheit versus physischer Abdeckung. GBS bewahrt oft die Tiefe pro Locus und den Populationsmaßstab, während Skim-Sequenzierung einen breiteren genomischen Kontext bewahrt, jedoch auf Kosten einer größeren Unsicherheit in sehr großen oder dosissensitiven Genomen.
Abbildung 2. Der eigentliche Kompromiss besteht nicht darin, "günstig versus umfassend", sondern in der Genotyp-Sicherheit versus physischer Abdeckung. GBS bewahrt oft die Tiefe pro Locus und den Populationsmaßstab, während Skim-Sequenzierung einen breiteren genomischen Kontext bewahrt, jedoch auf Kosten einer größeren Unsicherheit in sehr großen oder dosissensitiven Genomen.
Dieser Kompromiss erklärt auch, warum die Häufigkeit von Markern niemals ohne interpretativen Kontext berichtet werden sollte. Ein größeres Markerset ist nur dann besser, wenn seine Fehlerstruktur mit dem Vererbungssystem, das modelliert wird, kompatibel bleibt.
Die Häufigkeit von Markern ist nicht dasselbe wie die Souveränität von Markern.
Bei hochdichten Karten ist die rohe SNP-Zählung eine der am wenigsten zuverlässigen Zusammenfassungsmetriken. Ein kleinerer Markersatz mit stabilen Aufrufen, nützlichem Abstand und biologisch kohärenter Segregation kann einen viel größeren Katalog von schwachen, gruppierten oder dosis-ambigen Loci übertreffen.
Die Markerhoheit ergibt sich aus der Kontrolle über drei Dinge: wo Marker fallen, wie sicher sie genannt werden und ob das Artenmodell sie korrekt interpretieren kann. Ein Datensatz mit ungleicher physischer Verteilung kann dennoch gut funktionieren, wenn er die rekombinationsaktiven Segmente erfasst, die von Bedeutung sind. Ein Datensatz mit breiter physischer Reichweite kann jedoch scheitern, wenn die Tiefe zu gering ist, um vertrauenswürdige Genotypübergänge zu unterstützen.
Deshalb ist das Filtern von Daten in der Philosophie so wichtig. Gutes Filtern zielt nicht nur darauf ab, offensichtlich schlechte Loci zu entfernen. Es hat zum Ziel, die Teilmenge von Markern beizubehalten, deren Signal mit der Biologie der Art, dem Sequenzierungsdesign und dem letztendlichen Mapping-Modell kompatibel ist. In vielen Projekten wird diese Filterphase mit speziellen Strategien zur Marker-Generierung kombiniert, wie zum Beispiel Ganzgenom-SNP-Genotypisierung wenn der Schwerpunkt auf der Entdeckung dichter Polymorphismen vor der Kartenverfeinerung liegt.
Das nächste Problem ergibt sich direkt aus diesem Prinzip. Sobald die Art polyploid ist oder die Vererbung von klaren diploiden Annahmen abweicht, reicht die Markerqualität allein nicht mehr aus. Die Analyse muss auch bestimmen, wie viele Kopien jedes Allels vorhanden sind und wie diese Kopien über die homologen Chromosomen verteilt sind.
Die Alleldosierung ist der erste nicht verhandelbare Schritt.
In der polyploiden Kopplungsanalyse ist die Alleldosierung keine Verfeinerung. Sie ist die Eingangsbedingung für jeden späteren Inferenzschritt. Wenn die Dosierung falsch ist, wird die Phase instabil, die Rekombinationszahlen werden verzerrt, und die endgültige Karte beginnt, Genotypunsicherheiten so zu absorbieren, als ob es sich um echtes Chromosomenverhalten handeln würde.
Das Kernproblem ist einfach. In einem diploiden Organismus können viele Loci durch drei vertraute Zustände dargestellt werden: homozygot referenziell, heterozygot und homozygot alternativ. In einem tetraploiden Organismus kann dasselbe Locus in mehreren Allel-Kopien-Zuständen existieren. Eine alternative Kopie von vier ist nicht gleichwertig mit zwei von vier, und auch nicht mit drei von vier. Jeder Zustand hat eine andere Segregationserwartung. Wenn diese Zustände in eine generische heterozygote Klasse zusammengefasst werden, verliert die Karte die Erbstruktur, die sie benötigt, um Rekombination korrekt zu rekonstruieren.
Die Lesetiefe wird in diesem Stadium entscheidend. An einem biallelischen Locus kann das Verhältnis von Referenz- zu Alternativ-Lesungen einen ersten Hinweis auf die Dosisklasse geben. Theoretisch sollten sich die Cluster trennen. In der Praxis überlappen sie jedoch oft aufgrund von Stichprobenvariabilität, allelspezifischer Verzerrung, Mapping-Unschärfe, Wiederholungsinhalt und Verzerrungen auf Bibliotheksebene. Ein guter Workflow tut nicht so, als wären rohe Verhältnisse genau. Er behandelt die Dosisinferenz als ein Wahrscheinlichkeitsproblem und filtert Loci entsprechend dem Vertrauen statt nach wünschenswerter Präzision.
Deshalb kann die harte Genotypbestimmung in polyploiden Datensätzen riskant sein. Ein flacher oder grenzwertiger Locus kann nützlich sein, wenn die Unsicherheit ehrlich weitergegeben wird. Derselbe Locus wird schädlich, wenn er in eine feste Klasse gezwungen wird und dann als Beweis für einen Haplotype-Breakpoint interpretiert wird. In dichten Karten kann dieser Fehler die lokale Distanz erhöhen, falsche Rekombinationsevents erzeugen und die Reihenfolge benachbarter Marker destabilisieren.
Die praktische Regel ist klar. Dosierungsbewusste Genotypisierung muss vor dem aggressiven Kartenbau erfolgen, nicht danach. Loci sollten mit den erwarteten Segregationsmustern, den elterlichen Genotypen und der lokalen Konsistenz mit umliegenden Markern überprüft werden. Grenzwertige Loci sollten nicht immer verworfen werden, aber sie sollten nicht das gleiche interpretative Gewicht wie hochkonfidente Dosierungsaufrufe erhalten. In vielen Projekten mit komplexen Genomen beginnt der Unterschied zwischen einer stabilen Karte und einer aufgeblähten an diesem Schritt.
Hier beginnt auch die Verbindung zwischen der Wahl der Plattform und der nachgelagerten Genotypisierungsstrategie. Wenn umfassende Entdeckungsdaten nicht ausreichen, um unsichere Markerzustände zu stabilisieren, muss ein Projekt möglicherweise die Karte mit gezielter Bestätigung ergänzen durch Gezielte Regionssequenzierung oder hochkonfidente Lokusuntersuchung durch SNP-Fine-Mapping, insbesondere wenn wichtige Breakpoints oder Intervallgrenzen von einer relativ kleinen Anzahl entscheidender Marker abhängen.
Die Haplotyp-Phasierung in Polyploiden funktioniert am besten auf Blockebene.
Einzelne SNPs sind praktische analytische Einheiten, aber sie sind oft schwache biologische Einheiten. In komplexen Genomen, insbesondere bei Polyploiden, ist die bedeutungsvollere Frage nicht, welcher isolierte SNP seinen Zustand geändert hat, sondern welches vererbte Chromosomensegment seinen Zustand geändert hat. Deshalb schneiden Haplotypblöcke in der Regel besser ab als einzelne Marker als hauptsächliche Interpretationseinheit.
In einem Polyploid ist das Phasieren kein einfaches Buchhaltungsproblem mit zwei Chromosomen. Der genetische Kartierungsprozess muss mehrere Homologe verfolgen, deren Paarungsverhalten von der Art und dem Genomtyp abhängt. Bei Autopolyploiden kann die multisomische Vererbung flexible Paarungsbeziehungen zwischen den Homologen erzeugen. Bei Allopolyploiden kann die bevorzugte Paarung ein disomisches Muster schaffen, jedoch hängt die Unterscheidung der Homologen weiterhin davon ab, genügend dosisaufgelöste Markerinformationen zu haben, um subgenomische Segmente zuverlässig zu trennen.
Ein blockbasierter Ansatz verbessert die Stabilität auf zwei Arten. Erstens bündelt er Informationen über benachbarte Loci, was die Inferenz weniger empfindlich gegenüber Rauschen an einem einzelnen Marker macht. Zweitens entspricht er näher der meiotischen Realität. Rekombination verändert in der Regel die Vererbung auf Segmentebene, nicht auf der Ebene isolierter SNP-Umschaltungen. Wenn sich ein phasierter Block verschiebt, ist es viel wahrscheinlicher, dass dieses Ereignis eine echte Rekombinationsgrenze darstellt als ein einzelner inkonsistenter Marker.
Dies wird besonders wichtig in dichten Marker-Datensätzen, in denen die Anzahl der Marker die Anzahl der informativen Crossing-over-Ereignisse bei weitem übersteigt. Ohne Blocklogik häufen sich lokale Marker-Konflikte und zwingen die Karte zu unnötigen Mikroanpassungen. Mit blockbewusster Phasierung kollabieren die meisten dieser Konflikte in eine ehrlichere Zusammenfassung: Das Chromosom hat nicht genügend Beweise geliefert, um diese Loci einzeln zu trennen, daher sollten sie als Teil derselben vererbten Einheit interpretiert werden.
Dies ist auch ein Grund, warum langfristige Sequenzinformationen wertvoll werden können, sobald standardisierte Kurzlese-Marker die Struktur nicht mehr klar auflösen. In besonders schwierigen Intervallarchitekturen können ergänzende Daten von Nanopore Ultra-Lang Sequenzierung oder Telomer-zu-Telomer-Sequenzierung kann helfen, den strukturellen Kontext um unterdrückte Rekombinationsblöcke zu klären, insbesondere wenn physische Kontinuität relevant wird, um phasierte Intervalle zu interpretieren, anstatt lediglich SNPs aufzulisten.
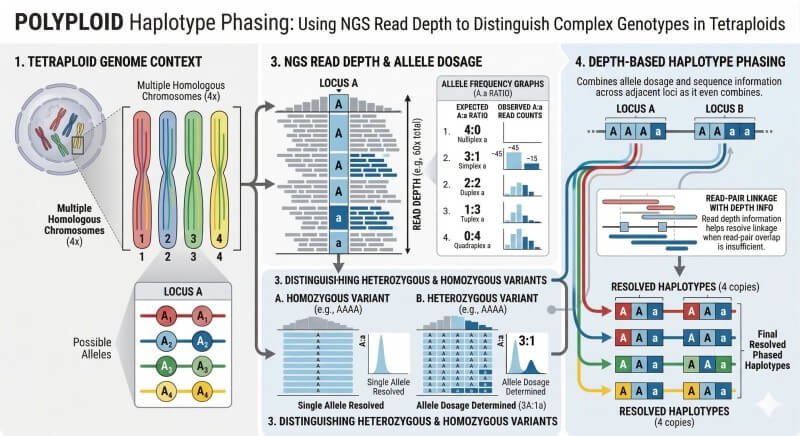 Abbildung 3. Die unterstützten Dosisklassen mit Lesetiefe werden am nützlichsten, wenn sie in phasierte Haplotypblöcke konsolidiert werden, da die Vererbung auf Blockebene stabiler ist als die Schwankungen einzelner Marker und die tatsächlichen Rekombinationsgrenzen in polyploiden Genomen genauer widerspiegelt.
Abbildung 3. Die unterstützten Dosisklassen mit Lesetiefe werden am nützlichsten, wenn sie in phasierte Haplotypblöcke konsolidiert werden, da die Vererbung auf Blockebene stabiler ist als die Schwankungen einzelner Marker und die tatsächlichen Rekombinationsgrenzen in polyploiden Genomen genauer widerspiegelt.
Eine starke Karte behandelt daher das Phasieren als ein segmentales Inferenzproblem. Das Ziel ist nicht, die Anzahl der individuell gekennzeichneten Marker zu maximieren. Es geht darum, zu rekonstruieren, welche homolog-verknüpften Blöcke übertragen wurden und wo die tatsächlich unterstützten Bruchpunkte liegen.
Die Bin-Mapping-Logik ist der Weg, wie dichte Markertabellen zu interpretierbaren Karten werden.
Das Bin-Mapping wird oft als ein praktischer Schritt zur Reduzierung der Markerüberlastung dargestellt. Tatsächlich ist es eine der klarsten Methoden, um die durch die Meiose auferlegte Informationsgrenze zu respektieren.
Die Argumentation ist einfach. Wenn eine Gruppe benachbarter Marker im gesamten Mapping-Population dasselbe Segregationsmuster zeigt, liefern diese Marker keine unabhängige Positionsinformation. Sie sind mehrere Messungen derselben durch Rekombination definierten Erbeeinheit. Wenn man sie alle als separat aufgelöste Punkte behandelt, schafft das visuelle Details, aber keine echte Auflösung.
Ein Bin erfasst das gemeinsame Signal und stellt es mit einer einzigen effektiven Einheit für die Bestellung dar. Dies verwirft keine nützliche Biologie. Es verwirft Redundanz. Der vollständige Satz von Markern innerhalb des Bins kann weiterhin für Annotationen, Genomprojektionen und die Interpretation von Kandidatenintervallen beibehalten werden. Was sich ändert, ist die Logik der Karte. Der Algorithmus wird aufgefordert, Rekombinationseinheiten zu sortieren, anstatt Tausende von nahezu identischen Beobachtungen.
Dies wird besonders nützlich in Regionen mit geringer Rekombination, starker Interferenz oder starker Marker-Sättigung. In diesen Segmenten kann das Erzwingen einer einzigartigen Reihenfolge unter ko-segregierenden Markern instabile lokale Anordnungen und künstliche Kartenexpansionen erzeugen. Binning verhindert dies, indem es die Kartenstruktur mit der Anzahl der Bruchpunkte in Einklang bringt, die die Population tatsächlich aufgedeckt hat.
Gutes Binning ist keine blinde Kompression. Über-Binning kann informative Breakpoint-Strukturen verbergen, wenn echte lokale Rekombinanten existieren. Unter-Binning bewahrt zu viel Redundanz und lässt kleine Genotypinkonsistenzen als bedeutungsvolle Struktur erscheinen. Das Ziel ist nicht maximale Vereinfachung. Es geht um eine proportionale Darstellung des tatsächlichen Rekombinationsinhalts des Datensatzes.
Ein starker Bin-Mapping-Workflow folgt oft vier Schritten. Zuerst werden Loci mit schlechtem Segregationsverhalten oder unakzeptabler Unsicherheit entfernt. Zweitens werden Marker identifiziert, die bei Individuen ko-segregieren oder nahezu ko-segregieren. Drittens werden Bins um gemeinsame Vererbungsmuster und verifizierte Übergänge an Bruchpunkten definiert. Viertens werden repräsentative Bin-Marker für den Kartenbau verwendet, während die vollständige Bin-Mitgliedschaft für spätere biologische Annotationen erhalten bleibt. Dies ergibt ein stabiles Rekombinationsgerüst, ohne die nachgelagerte Vielfalt zu opfern.
Diese gleiche Logik wird noch mächtiger, wenn Markersysteme absichtlich um die Auflösungsregelung von Vererbung herum gestaltet werden, anstatt sich nur auf die reine Anzahl der Marker zu stützen. Ansätze wie ddRAD-seq, 2b-RADoder Multiplex-PCR-Sequenzierung kann jeweils unterschiedliche Muster von Marker-Dichte, lokaler Redundanz und Sichtbarkeit von Bruchpunkten erzeugen. Die richtige Wahl hängt weniger von der Durchsatzrate der Überschrift ab, als vielmehr davon, ob die resultierenden Marker sauber in rekombinationsunterstützte Bins zusammengefasst werden können.
Von der QTL-Erkennung zur Feinkartierung
Der Übergang von der Verknüpfungskarte zur QTL-Analyse sieht in Arbeitsabläufen und Abbildungen oft unkompliziert aus. In realen Datensätzen ist dies der Punkt, an dem viele Projekte herausfinden, ob die Karte tatsächlich verwendbar ist. Breite QTL-Erkennung kann einige lokale Unsicherheiten tolerieren. Feinkartierung kann das nicht.
Ein erster QTL-Scan ist darauf ausgelegt, Chromosomenregionen zu finden, die mit der Variabilität von Merkmalen assoziiert sind. In einem dichten Markersatz können diese Regionen täuschend präzise erscheinen, da die Markerabdeckung visuell intensiv ist. Aber die Marker-Dichte ist nicht dasselbe wie rekombinante Diversität. Ein scharf aussehender Gipfel kann sich immer noch innerhalb eines breiten Vererbungblocks befinden, der zu wenige informative Bruchpunkte aufweist, um ein minimales Intervall mit Zuversicht zu isolieren.
Deshalb ist Fine-Mapping nicht einfach eine Frage des Hinzufügens weiterer Marker. Es hängt davon ab, die richtige Struktur in der ursprünglichen Karte zu haben: stabile Dosierungsaufrufe, glaubwürdige Phasenbeziehungen, sinnvolle Bins und ein realistisches Verständnis dafür, wo Rekombination tatsächlich informativ ist. Wenn diese Struktur schwach ist, verengt dichtere Genotypisierung oft den Intervall kosmetisch statt biologisch.
Eine disziplinierte Feinkartierungsstrategie stützt sich normalerweise auf zwei Formen der Verfeinerung. Die erste ist die strukturelle Verfeinerung: die Stabilisierung der Karte, sodass die Rekombinationsgrenzen vertrauenswürdig sind. Die zweite ist die inferentielle Verfeinerung: die Verwendung von Modellen, die lokale Signale von Hintergrundgenetik-Effekten trennen und die Aufmerksamkeit auf die informativsten Rekombinanten konzentrieren.
Dieser zweite Schritt ist der Punkt, an dem viele Projekte entweder vorankommen oder ins Stocken geraten. Wenn nur eine kleine Anzahl informativer Rekombinanten innerhalb des Zielintervalls existiert, wird keine noch so große rechnerische Verfeinerung eine echte kausale Auflösung schaffen. In solchen Fällen kann der beste nächste Schritt darin bestehen, die Population zu erweitern, gezielt Individuen mit Bruchpunkten auszuwählen oder die Region mit gezielteren Tests zu ergänzen. Für die gezielte Nachverfolgung des Intervalls, Amplicon-Sequenzierungsdienste oder Gezielte Regionssequenzierung kann nützlicher sein als einfach einen genomweiten Test mit demselben Unsicherheitsgrad zu wiederholen.
Die komposite Intervallkartierung verbessert die Auflösung nur, wenn die Karte bereits glaubwürdig ist.
Die komposite Intervallkartierung bleibt relevant, da die Merkmalsvariation selten von einem isolierten Chromosomenabschnitt kontrolliert wird. Hintergrundloci tragen zur Varianz bei. Verknüpfte Regionen können sich gegenseitig verwischen. Dichte Markersets können breite Gipfel erzeugen, die stark erscheinen, während sie dennoch schwer zu analysieren sind.
CIM hilft, indem es Hintergrundmarker als Cofaktoren einführt, während das fokale Intervall bewertet wird. Diese Cofaktoren absorbieren einen Teil der Variation, die von anderen genomischen Regionen beigetragen wird, was oft das lokale QTL-Profil schärft und die Trennung zwischen nahen Signalen verbessert. In einem gut strukturierten Datensatz kann dies Verzerrungen reduzieren und die Interpretation von Effekt-Schätzungen erleichtern.
Aber CIM ist kein Reparaturwerkzeug für eine schwache Karte. Wenn die Markerreihenfolge instabil ist oder die Dosierungsunsicherheit ungelöst bleibt, kann die Auswahl von Kofaktoren die Artefaktstruktur anstelle der tatsächlichen Hintergrundvarianz absorbieren. Wenn die zugrunde liegende Karte durch falsche Bruchpunkte oder verzerrte Phasenübergänge aufgebläht ist, kann CIM das falsche Signal schärfen und die Ausgabe sicherer erscheinen lassen, als sie tatsächlich ist.
Eine nützliche Regel ist einfach: CIM ist am wertvollsten, nachdem das Rekombinationsgerüst bereits vertrauenswürdig ist. Wenn die Population klare phasenunterstützte Segmente, kohärente Bins und eine stabile lokale Anordnung unter sinnvollen Filteränderungen zeigt, kann CIM den Intervallkontrast verbessern. Fehlen diese Bedingungen, sollte das Projekt die Kartenstruktur reparieren, bevor ein Kofaktormodell zur Verfeinerung herangezogen wird.
In einigen Arbeitsabläufen umfasst dieser Reparaturschritt auch einen stärkeren strukturellen Kontext. Zum Beispiel, wenn lokale Intervallambiguität ungelöste chromosomale Anordnungen widerspiegelt, anstatt einfaches Markerrauschen, integriert Hi-C-Sequenzierung oder sogar de novo Genomressourcen wie Pflanzen-/Tier-Whole-Genome de novo Sequenzierung kann mehr zur Verbesserung der Intervallglaubwürdigkeit beitragen als eine weitere Runde rein statistischer Anpassungen.
Feinabgleich funktioniert am besten, wenn Haplotypen isolierte Marker ersetzen.
Die nützlichsten Feinkartierungsintervalle werden oft nicht durch einen einzelnen Marker definiert, sondern durch ein kurzes vererbtes Haplotypsegment, das mit dem Phänotyp über informative Rekombinanten hinweg assoziiert bleibt. Dies ist ein stärkeres und realistischeres Ziel.
Ein einzelner SNP kann die Region markieren, aber der tatsächliche biologische Unterschied kann mehrere verknüpfte Varianten, ein regulatorisches Segment, ein strukturelles Merkmal oder einen haplotypenspezifischen Zustand des Subgenoms umfassen. Haplotype-bewusstes Feinkartieren ist besser geeignet für diese Realität, da es verfolgt, welches vererbte Segment mit dem Phänotyp gekoppelt bleibt, während benachbarte Segmente durch Rekombination getrennt werden.
In der Praxis bedeutet dies, phasierte Blöcke, verifizierte Breakpoint-Positionen und Merkmalsmuster zu überlagern, um das kleinste erhaltene Segment zu identifizieren, das das Signal weiterhin erklärt. Die Qualität dieses Ergebnisses hängt von jeder vorherigen Entscheidung ab: Plattformauswahl, Dosismodellierung, Phasendisziplin, Bin-Konstruktion und hintergrundbewusste Intervallanalyse. Feinabstimmung ist kein separater Akt am Ende. Es ist die Belohnung dafür, die frühere Kartenarchitektur richtig zu gestalten.
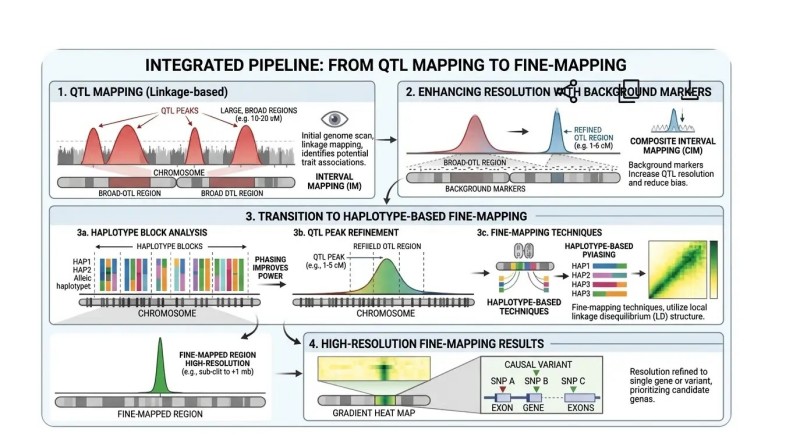 Abbildung 4. Breite QTL-Spitzen werden biologisch nur dann enger, wenn die durch Rekombination unterstützte Struktur erhalten bleibt, Hintergrundeffekte kontrolliert werden und das endgültige Intervall als ein beibehaltener Haplotypabschnitt und nicht als ein Einzelmarker-Spike interpretiert wird.
Abbildung 4. Breite QTL-Spitzen werden biologisch nur dann enger, wenn die durch Rekombination unterstützte Struktur erhalten bleibt, Hintergrundeffekte kontrolliert werden und das endgültige Intervall als ein beibehaltener Haplotypabschnitt und nicht als ein Einzelmarker-Spike interpretiert wird.
Dies ist auch der Grund, warum die letzten Phasen der Intervallverfeinerung oft von einer gestuften Prüfstrategie profitieren. Breite Entdeckungsmethoden helfen dabei, Kandidatenregionen zu identifizieren, aber eine hochgradige Eingrenzung hängt normalerweise von einer gezielteren Validierung ab. In vielen Projekten, SNP-Fine-Mapping wird zur natürlichen Brücke zwischen einem Verknüpfungssignal und einem besser verteidigbaren minimalen Intervall.
Ein vertrauenswürdiger Intervallrahmen ist wichtiger als eine lange Markerliste.
Der wahre Wert einer Verknüpfungskarte liegt nicht in der Gesamtzahl der SNPs oder der gesamten Zentimorgan-Länge. Es geht darum, ob die Karte einen vertrauenswürdigen Intervallrahmen für die biologische Interpretation unter realen Rekombinationsbedingungen bietet.
Ein vertrauenswürdiges Framework hat erkennbare Eigenschaften. Seine Abstände werden nicht offensichtlich durch Genotypfehler aufgebläht. Seine lokale Ordnung bleibt unter sinnvollen Filteränderungen stabil. Seine Dosierungszuweisungen entsprechen dem Vererbungssystem der Art. Seine dichten Marker werden dort, wo es nötig ist, in recombination-unterstützte Einheiten zusammengefasst. Seine QTL-Intervalle verengen sich aufgrund informativer Bruchpunkte, nicht weil die Markerhäufigkeit falsche Präzision erzeugt.
Das ist der praktische Standard für die Verknüpfungsanalyse komplexer Genome. In großen Genomen und Polyploiden kommt Präzision nicht nur von der Dichte. Sie ergibt sich aus der Berücksichtigung von Crossing-over-Interferenz, der Auswahl von Markersystemen entsprechend dem tatsächlichen Engpass des Projekts, der ehrlichen Modellierung der Dosis, dem Phasieren auf Blockebene, dem Binning redundanter Marker und der Verwendung von Intervallmethoden nur, wenn das Kartenrückgrat stabil ist. Wenn diese Bedingungen erfüllt sind, wird die Verknüpfungsanalyse mehr als nur eine Anordnungsübung. Sie wird zu einem zuverlässigen Rahmen für Entdeckungen auf Intervallniveau.
Häufig gestellte Fragen
Was ist der größte Fehler in der hochdichten Verknüpfungsanalyse?
Der häufigste Fehler besteht darin, anzunehmen, dass mehr Marker automatisch eine bessere Auflösung bedeuten. In Wirklichkeit hängt die Auflösung von informativen Rekombinationsevents ab, nicht nur von der Anzahl der Marker. Wenn die Markerdichte die lokale Bruchpunktdichte erheblich übersteigt, kann die Karte sehr detailliert erscheinen, während sie strukturell schwach bleibt. Deshalb sind Co-Segregationslogik, Bin-Konstruktion und phasenbewusste Interpretation oft wichtiger als das Hinzufügen einer weiteren Schicht von SNP-Häufigkeit.
Wann sollte Kosambi Haldane vorgezogen werden?
Kosambi ist in der Regel geeigneter, wenn erwartet wird, dass Crossover-Interferenz von Bedeutung ist, da es nicht-zufällige Abstände zwischen Crossover-Ereignissen annimmt. Haldane ist nützlich, wenn ein Modell ohne Interferenz getestet oder als Benchmark verwendet wird. Die stärkere Praxis besteht darin, die Sensitivität über Funktionen hinweg zu vergleichen, anstatt eine von beiden als automatischen Standard zu behandeln.
Wie sollten Forscher GBS im Vergleich zu Low-Pass-Whole-Genome-Skim-Sequenzierung betrachten?
Die Wahl sollte gemäß dem schwächsten Punkt im Studiendesign getroffen werden. GBS funktioniert oft besser, wenn die Stichprobengröße die Hauptbeschränkung darstellt und eine erste Verknüpfungskarte das Ziel ist. Low-Pass-Skim-Sequenzierung wird attraktiver, wenn ein breiterer Genomkontext, die Wiederverwendung von Scaffolds oder die spätere Haplotyp-Interpretation von Bedeutung sind. In dosis-sensitiven Systemen sollte die Plattform, die keine stabile Allel-Zustandsinferenz bewahren kann, zuerst abgelehnt werden.
Warum ist die Alleldosierung in der Tetraploidenkartierung so wichtig?
Da ein tetraploider Locus in mehreren Allel-Kopienzuständen existieren kann und diese Zustände sich nicht auf die gleiche Weise segregieren. Wenn sie in diploide Aufrufe zusammengefasst werden, verliert die Karte kritische Erbinformationen. Dosierungsfehler sind besonders schädlich, da sie falsche Breakpoint-Signale erzeugen und sowohl die lokale Phase als auch die gesamte Kartenlänge verzerren können.
Was löst die Bin-Zuordnung, das gewöhnliche dichte Karten nicht tun?
Es löst das Redundanzproblem. Wenn viele benachbarte Marker das gleiche Vererbungsmuster zeigen, bieten sie keine unabhängige Anordnungsinformation. Die Bin-Kartierung fasst sie in recombination-unterstützte Einheiten zusammen, was die Markerreihenfolge stabilisiert und die künstliche Kartenerweiterung reduziert, ohne das Potenzial für nachgelagerte Annotationen zu opfern.
Warum bleibt die komposite Intervallkartierung relevant?
Da dichte Marker-Datensätze weiterhin Hintergrundgenetik-Effekte und verknüpfte Störungen enthalten, kann CIM die QTL-Auflösung verbessern, indem es Hintergrundloci berücksichtigt, während das fokale Intervall getestet wird. Es funktioniert jedoch nur gut, wenn die zugrunde liegende Karte bereits stabil ist. Es schärft die glaubwürdige Struktur; es schafft keine Glaubwürdigkeit aus instabiler Marker-Architektur.
Kann eine Verknüpfungskarte die Verbesserung der Genomassemblierung unterstützen?
Ja. Eine stabile Verknüpfungskarte kann helfen, Gerüste zu verankern, die langfristige Anordnung zu validieren und strukturelle Inkonsistenzen in einer Assemblierung zu identifizieren. Dies ist besonders nützlich bei Nicht-Modellarten oder großen Genomen, bei denen eine sequenzbasierte Assemblierung allein möglicherweise nicht mit Zuversicht die chromosomale Anordnung erfasst.
Referenzen
- Haldane JBS. Die Kombination von Verknüpfungswerten und die Berechnung der Abstände zwischen den Loci verknüpfter Faktoren. Zeitschrift für Genetik. 1919;8:299–309. DOI: 10.1007/BF02983075
- Kosambi DD. Die Schätzung von Kartenentfernungen aus Rekombinationswerten. Annalen der Eugenik. 1944;12:172–175. DOI: 10.1111/j.1469-1809.1943.tb02321.x
- Lander ES, Botstein D. Kartierung mendelnder Faktoren, die quantitativen Merkmalen zugrunde liegen, mithilfe von RFLP-Verknüpfungskarten. Genetik. 1989;121(1):185–199. DOI: 10.1093/genetics/121.1.185
- Zeng Z-B. Präzise Kartierung von quantitativen Merkmalsloci. Genetik. 1994;136(4):1457–1468. DOI: 10.1093/genetics/136.4.1457
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. Ein robuster, einfacher Genotypisierungs- durch Sequenzierung (GBS) Ansatz für Arten mit hoher Diversität. PLOS ONE. 2011;6(5):e19379. DOI: 10.1371/journal.pone.0019379
- Rastas P. Lep-MAP3: robuste Verknüpfungsanalyse selbst für niedrig-qualitative Whole-Genome-Sequenzierungsdaten. Bioinformatik. 2017;33(23):3726–3732. DOI: 10.1093/bioinformatics/btx494
- Bourke PM, van Geest G, Voorrips RE, Jansen J, Kranenburg T, Shahin A, Visser RGF, Arens P, Smulders MJM, Maliepaard C. polymapR—Verknüpfungsanalyse und genetische Kartenkonstruktion aus F1-Populationen von auskreuzenden Polyploiden. Bioinformatik. 2018;34(20):3496–3502. DOI: 10.1093/bioinformatics/bty371
- Mollinari M, Garcia AAF. Verknüpfungsanalyse und Haplotyp-Phasierung in experimentellen Autopolyploid-Populationen mit hohem Ploidiegrad unter Verwendung versteckter Markov-Modelle. G3: Gene, Genome, Genetik. 2019;9(10):3297–3314. DOI: 10.1534/g3.119.400378
- Mollinari M, Olukolu BA, Pereira GS, Khan A, Gemenet D, Yencho GC, Zeng Z-B. Entschlüsselung der hexaploiden Süßkartoffelvererbung mittels ultra-dichter Multilokus-Kartierung. G3: Gene, Genome, Genetik. 2020;10(1):281–292. DOI: 10.1534/g3.119.400620
- Han K, Jeong HJ, Yang HB, Kang SM, Kwon JK, Kim S, Choi D, Kang BC. Eine ultra-hochdichte Bin-Karte erleichtert das Hochdurchsatz-QTL-Mapping von gartenbaulichen Merkmalen in Paprika. DNA-Forschung. 2016;23(2):81–91. DOI: 10.1093/dnares/dsw001
- Shirasawa K, Hirakawa H, Nunome T, Tabata S, Isobe S. Eine hochdichte SNP-Genkarte, die aus einem vollständigen Satz homologer Gruppen in autohexaploidem Süßkartoffel (Ipomoea batatas) besteht. Wissenschaftliche Berichte. 2017;7:44207. DOI: 10.1038/srep44207
- Stift M, Berenos C, Kuperus P, van Tienderen PH. Segregationsmodelle für disomische, tetrasomische und intermediate Vererbung in Tetraploiden: ein allgemeines Verfahren, das auf Hybriden von tetraploiden Ranunculus-Arten angewendet wird. Genetik. 2008;179(4):2113–2123. DOI: 10.1534/genetics.107.085027
Verwandte Dienstleistungen
Nur für Forschungszwecke. Nicht für den Einsatz in diagnostischen Verfahren.


