
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
eccDNA-Sequenzierung erklärt: Was sie misst und warum Forscher sie verwenden (RUO)
Anfänger in der biomedizinischen Forschung erleben denselben Wandel, den viele Genomlabore im vergangenen Jahrzehnt gespürt haben: Das Genom ist nicht nur eine Ansammlung von langen, ordentlichen Chromosomen. Zellen tragen auch vielfältige extrachromosomale zirkuläre DNA—eccDNAs—die unerwartete Schichten von Variation und Regulation hinzufügen. Wenn Sie nach "eccdna nature" gesucht haben, um zu verstehen, warum Kreise in hochkarätigen Veröffentlichungen immer wieder auftauchen, wird Ihnen dieser Leitfaden einen klaren, praktischen Ausgangspunkt bieten.
EccDNA (extrachromosomale zirkuläre DNA) bezieht sich auf zirkuläre DNA-Moleküle, die außerhalb der chromosomalen DNA im Zellkern (und gelegentlich in zytoplasmatischen Fraktionen) gefunden werden. Sie unterscheiden sich von Plasmiden (gewöhnlich exogen, vektorbasierte Zirkeln in Laborsystemen) und mitochondrialer DNA (mtDNA, ein endogenes zirkuläres Genom, das auf Mitochondrien beschränkt ist). Im Rahmen von eccDNA reservieren Forscher oft "ecDNA" für große, hoch amplifizierte Zirkeln, die häufig in Krebs vorkommen und intakte Onkogene tragen. Die Übersichten konvergieren auf diese Einordnung: kleine eccDNAs sind in Geweben weit verbreitet, während ecDNA (häufig >~1 Mb) überwiegend tumorassoziiert und mit Amplifikation und Heterogenität verbunden ist, wie von Wang (2024) in einer Open-Access-Übersicht dargelegt und durch aktuelle Zusammenfassungen der ecDNA-Eigenschaften in Krebs bekräftigt. Siehe die detaillierten Unterscheidungen in der Open-Access-Synthese von Wang in "Die Geheimnisse der extrachromosomalen zirkulären DNA enthüllen" (2024) und Zhou's Diskussion der ecDNA-Eigenschaften in "Frontiers in Genetics" (2024).
- Laut der Open-Access-Übersicht von Wang (2024)eccDNAs reichen von Mikro-DNA (~200 bp–3 kb) bis zu größeren Elementen, während ecDNA typischerweise viel größere Zirkeln bezeichnet, die oft ganze Gene enthalten.
- Zhou (2024) in Frontiers in Genetics betont, dass ecDNA in Tumoren weit verbreitet ist, im Gegensatz zu kilobasengroßen eccDNAs, die in verschiedenen Kontexten vorkommen.
Schnelles Glossar (für Anfänger)
- eccDNAOberbegriff für extrachromosomale zirkuläre DNA unterschiedlicher Größen und Ursprünge; häufig in verschiedenen Geweben und Bedingungen anzutreffen.
- ecDNAGroße (>~100 kb bis Mb-Skala) zirkuläre DNA in Krebs, die häufig Onkogen-Amplifikationen trägt und die Genexpression verstärkt.
- MikroDNAKleine eccDNA (Hunderte bis einige tausend Basenpaare) sind oft in der Nähe von genreichen oder repetitiven Regionen angereichert.
- ERCs (episomale rDNA-Zirkel)Kreise, die aus ribosomaler DNA abgeleitet sind, klassisch in der Alterung von Hefen untersucht.
- KreisverkehrDer Fusionspunkt, an dem ein linearer DNA-Fragmente sich selbst wieder verbindet, um einen Kreis zu bilden; nachgewiesen durch gespaltene/inkongruente Reads in der Sequenzierung.
Abbildung 1: Einfache Vergleich von DNA-Formen — lineare chromosomale DNA (blau #0099d8), extrachromosomale zirkuläre DNA (eccDNA; grün #82bc24) und mitochondriale DNA (orange). Größenlegende (nicht maßstabsgetreu): Chromosomale ≫ eccDNA (~100s bp–kb) ≈ mtDNA (~14–20 kb).
Alt-Text: Drei-Symbol-Schema: blaues Band für lineare chromosomale DNA, kleine grüne Kreise für eccDNA mit einem beschrifteten Verbindungs-Pfeil und ein orangefarbener Kreis innerhalb eines Mitochondriums für mtDNA.
Was eccDNA-Sequenzierung misst
EccDNA-orientierte Sequenzierung konzentriert sich auf Beweise, die standardmäßig Whole-Genome-Sequenzierung (WGS) reinigt sich nicht sauber: die Anwesenheit und Eigenschaften von Kreisen, die über die Chromosomen hinaus existieren. Praktisch messen Sie drei zentrale Dinge.
- Junktionsstruktur (wie der Kreis gebildet wird) Denken Sie an eine Kreisverbindung als den zentralen Punkt, an dem ein Fragment zurückfaltet und sich selbst wieder verbindet. In Kurzlesedaten sehen Sie geteilte Reads, die über die Fusion hinweg abgebildet sind, und gepaarte Reads, deren Orientierung auf nach außen gerichtete Junktionen hindeutet. In Langlesedaten können Sie manchmal den gesamten Kreis durchqueren. ATAC-seq—die Profile offenen Chromatins—können auch Junctions ohne zirkelspezifische Anreicherung erkennen, indem sie diese Mapping-Signaturen nutzen. Kumar und Kollegen zeigten 2020, dass ATAC-seq-Signaturen eccDNA-Junctions vorhersagen, die durch inverse PCR und FISH in Krebsmodellen validiert werden können. Siehe den methodologischen Kontext in Kumar et al. (2020) in Science Advances und die Open-Access-Erklärungsmaterialien in die entsprechenden Preprint-/PMC-Ressourcen.
- Geninhalt (welche Gene und Elemente getragen werden) Kreise können gesamte Gene, Genfragmente, Enhancer, Promotoren und Wiederholungen tragen. Der Geninhalt ist wichtig, da er auf die Funktion hinweist: ecDNA enthält häufig intakte Onkogene (z. B. EGFR, MYC, MYCN) und regulatorische Sequenzen, die die Transkription erhöhen. Kleine eccDNAs können lokale genomische Plastizität, Wiederholungsbiologie oder Stressreaktionen widerspiegeln. Für eine zugängliche Synthese von Rollen und Krankheitsassoziationen siehe Zhao (2022) in eLife und die Landschaftsübersicht von Zeng (2022) in Genome Research (Open Access).
- Kopienzahl und Häufigkeit (wie viel kreisförmiger Inhalt vorhanden ist) Schätzungen der Kopienzahl erfassen die Amplifikation unabhängig von der chromosomalen Lage. Bei Krebs können ecDNA-Amplikons hohe Kopienzahlen erreichen, ungleichmäßig während der Zellteilung segregieren und Heterogenität sowie Therapieresistenz antreiben. Im einfachen Sprachkontext beschreibt das NCI Cancer Genomics-Programm ecDNA als frei schwebende Schleifen, die Onkogene verstärken und zur Variabilität zwischen Zellen beitragen. Siehe NCI CCGs ecDNA-Übersicht (Blog von 2022) für eine lesbare Zusammenfassung.
Wenn Sie einen tieferen Einblick erhalten möchten, wie Proben isoliert und angereichert werden, bevor sie sequenziert werden, springen Sie zu unserem Schritt-für-Schritt-Leitfaden: Experimenteller Workflow für eccDNA-Sequenzierung.
Kernforschungsanwendungen
Warum verwenden Forscher eccDNA-SequenzierungZwei Anwendungsbereiche dominieren das Interesse und das Suchverhalten: Krebsbiologie und Alterung/Stress.
Krebsbiologie: Onkogenamplifikation und Heterogenität
Große ecDNA-Zirkel tragen häufig Onkogene wie EGFR, MYC, MYCN und CCND2. Da sie von Chromosomen getrennt sind, können sie hohe Kopienzahlen erreichen und ungleichmäßig segregieren, was zu Unterschieden zwischen den Zellen führt, die die Therapie komplizieren. Mechanistische Arbeiten zeigen, dass die Transkription von ecDNA die chromosomalen Gegenstücke übertreffen kann, unterstützt durch Enhancer-Hijacking und expansive Chromatin-Kontakte. Eine umfassende Übersicht von Yi und Kollegen aus dem Jahr 2022 fasst wiederkehrende Onkogen-Loci auf ecDNA in mehreren Tumorarten zusammen, während zugängliche Übersichten des NCI Heterogenität und Resistenzen hervorheben.
Echte Vignetten helfen, die Konzepte zu verankern:
- Glioblastom (GBM): EGFR-Amplifikation ist ein klassisches Beispiel für ecDNA im GBM, bei dem ungleiche Segregation mosaikartige Ausdrucks- und Reaktionsmuster erzeugt; ATAC-seq kann frühe Junction-Signaturen aufdecken, bevor die vollständige Amplifikation offensichtlich wird. Kumars Studie liefert die grundlegende Mapping-Logik: Wissenschaftliche Fortschritte (2020).
- Kleinzelliges Lungenkarzinom (SCLC): MYC/MYCN ecDNA-Einheiten zeigen, wie enhancer-reiche Amplifikationen die Transkription über chromosomale Vergleiche hinaus erhöhen und zu aggressiven Phänotypen beitragen, wie in Übersichtsarbeiten zusammengefasst. Yi (2022).
Entdecken Sie, wie diese Kreise die Tumorheterogenität in unserem Serienartikel vorantreiben: eccDNA bei Krebs.
Altern und Stress: Akkumulation und zelluläre Seneszenz-Kontexte
EccDNA hat eine lange Geschichte im Alterungsprozess von Hefezellen: ERCs sammeln sich in Mutterzellen an und tragen über Replikations- und Transkriptionseffekte zur Seneszenz bei. Die Beweise bei Säugetieren sind nuanciert – eccDNAs sind in normalen und krebsartigen Geweben vorhanden, und in einigen Kontexten zeigen sie unterschiedliche Profile im Zusammenhang mit Alterung oder Stress. Wangs Übersicht von 2024 fasst Subtypen und potenzielle Funktionen zusammen, während Zengs Landschaft von 2022 die Verteilungen über verschiedene Gewebe hervorhebt. Im Plasma zeigt eccDNA eine charakteristische Größenperiodizität im Vergleich zu linearem cfDNA, was mit den Effekten der nukleosomalen Verpackung übereinstimmt, wie von Sin und Kollegen berichtet.
- Hefe ERCs: Klassische Modelle zeigen die Ansammlung von ERCs und altersassoziierte Phänotypen in Mutterzellen; siehe moderne Zusammenfassungen in DNA-Zirkel fördern das Altern von Hefen (eLife, 2022) und Transkriptionsinduzierte Bildung von eccDNA in Hefe (PLoS Biology, 2019).
- Plasma/cfDNA: Deutliche eccDNA-Größenpeaks (~202 bp, ~338 bp) im Vergleich zur 166 bp modal linearen cfDNA, was den Chromatin-Kontext widerspiegelt; siehe Sin (2020) in PNAS.
Untersuchen Sie die Rolle von zirkulärem DNA in der zellulären Seneszenz und somatischen Biologie in unserem Serienartikel: eccDNA in somatischen Zellen und Alterung.
Methodenübersicht: Wie sequenzieren wir Kreise?
Es gibt kein einzelnes "eccDNA-Kit", das für jede Frage geeignet ist. Die meisten Labore wählen aus drei übergeordneten Ansätzen, die oft zur Validierung kombiniert werden.
- Exonuklease-Digestion + Rolling Circle Amplification (RCA): ATP-abhängige Exonukleasen (z. B. Plasmid-Safe) entfernen lineare DNA; phi29-Polymerase amplifiziert zirkuläre Vorlagen. Dieses Grundgerüst bildet die Grundlage vieler Circle-Seq-Workflows. Es ist effizient für kleine Kreise, kann jedoch gegen größere Kreise voreingenommen sein und kann Konkatemer-Artefakte erzeugen, wenn die Bedingungen nicht optimiert sind. Praktische Einblicke und Protokollgerüste erscheinen in Møller (2020) beschreibt Circle-Seq. und Synthesestücke wie Yu (2023) zu Circle-Seq-Varianten.
- Tn5/tagmentationsbasierte Ansätze: Circulome-seq und ATAC-seq nutzen die Tn5-Transposition, um zugängliche DNA zu kennzeichnen und Junction-Signaturen ohne RCA zu erfassen. Sie reduzieren einige Amplifikationsverzerrungen, können jedoch eine höhere Sequenzierungstiefe und sorgfältige bioinformatische Filterung erfordern. Siehe Kumar (2020) in Science Advances und die darin verlinkten Open-Access-Materialien.
- Nicht-RCA-Anreicherung für native kleine Zirkeln: Einige Arbeitsabläufe reichern kleine Zirkeln über Exonuklease-Reinigung ohne RCA an, wodurch die nativen Größenverteilungen erhalten bleiben und Chimären reduziert werden. Vergleichende Leistungsdiskussionen erscheinen in aktuellen Methodensynthesen wie Gao (2024) zu den Handels-offs bei der Rekonstruktion und Anreicherung von Langsequenzen.
Wichtige Planungsüberlegungen (anfängerfreundlich)
- Probenvorbereitung: Die Qualität der Kernisolierung beeinflusst die Effizienz der linearen DNA-Depletion; sanfte Handhabung reduziert das Scheren, das die Junction-Anrufe verfälschen kann.
- Kontrollen: Fügen Sie Spike-in-Plasmide hinzu, um die Verhältnisse von zirkulär zu linear zu verfolgen; quantifizieren Sie die mtDNA-Rückstände; fügen Sie Kontrollen für die Scheinverdauung hinzu, um unvollständige Exonuklease-Reinigung zu erkennen.
- Bibliothekskonstruktion: Für RCA-abgeleitetes Material die Amplifikationszyklen begrenzen, um Konkatemere zu reduzieren; für ATAC-seq die Transposition optimieren, um Sensitivität und Spezifität auszubalancieren.
- Sequenzierungsstrategie: Kurzzeit-Illumina ist gängig für Circle-Seq; fügen Sie Langzeit-ONT/PacBio hinzu, wenn Sie große Kreise oder komplexe Umstellungen erwarten.
Für Strategiedifferenzen und einzubeziehende Kontrollen vergleichen Sie die Optionen in unserem Serienartikel: Auswahl von eccDNA-Anreicherungsmethoden.
- Für Labore, die eine ausgelagerte RUO-Option bevorzugen, können Dienstleister Exonuklease-Digestion plus RCA durchführen und Berichte über Junction und Geninhalt zurückgeben; siehe CD Genomics. Onkologie-Forschungslösungen für die Studienplanung und Unterstützung bei der RUO-Sequenzierung.
Offenlegung: CD Genomics ist unser Produkt. Neutrales Beispiel für die Durchführung von Dienstleistungen: Anbieter wie CD Genomics kann RUO eccDNA-Workflows (z. B. Exonuklease-Digestion plus RCA) durchführen und Berichte über Junction- und Geninhalt mit angemessener Qualitätskontrolle liefern, wenn Projekte externe Unterstützung benötigen. Hier nur zur Kontextualisierung erwähnt.
 Abbildung 1: Konzeptschema. Chromosomales DNA ist lang und linear; eccDNAs sind vielfältige kleine Kreise, die oft von chromosomalen Fragmenten abgeleitet sind; mtDNA ist ein separates zirkuläres Genom in Mitochondrien.
Abbildung 1: Konzeptschema. Chromosomales DNA ist lang und linear; eccDNAs sind vielfältige kleine Kreise, die oft von chromosomalen Fragmenten abgeleitet sind; mtDNA ist ein separates zirkuläres Genom in Mitochondrien.
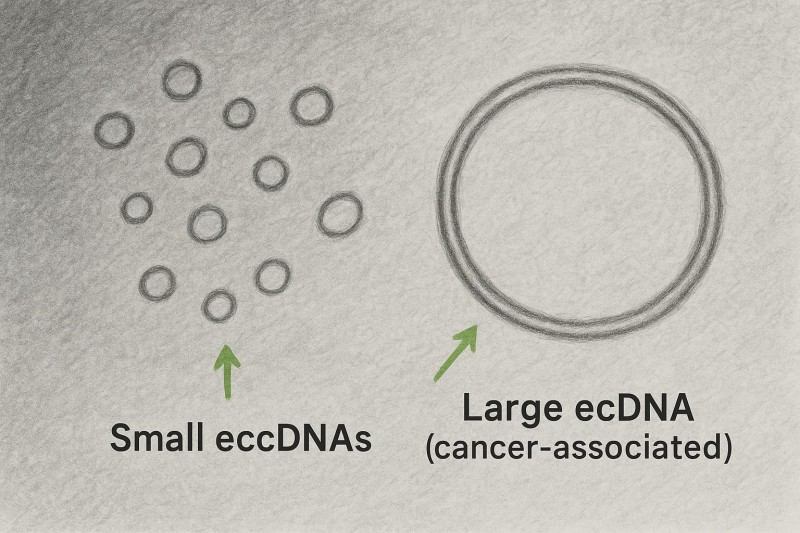 Abbildung 2: Stilisiertes EM/AFM-inspiriertes Zeichnung. Kleine eccDNAs erscheinen als dünne Schlaufen; eine größere Schlaufe repräsentiert krebsassoziiertes ecDNA. Dies ist eine illustrative Skizze, kein Mikrogramm.
Abbildung 2: Stilisiertes EM/AFM-inspiriertes Zeichnung. Kleine eccDNAs erscheinen als dünne Schlaufen; eine größere Schlaufe repräsentiert krebsassoziiertes ecDNA. Dies ist eine illustrative Skizze, kein Mikrogramm.
Praktische Planung für Anfänger (RUO)
Um die Konzepte greifbar zu machen, sind hier zwei Mini-Projekte und eine Fehlersuche-Checkliste, die Anfänger für Forschungszwecke anpassen können.
Mini-Projekt 1: Pilotprofilierung in einer Krebszelllinie
- Ziel: Erkennung von ecDNA, die ein Onkogen (z.B. EGFR) in einer Glioblastomlinie trägt.
- Route: Bereiten Sie Nuclei vor, führen Sie eine Exonuklease-Digestion durch, um lineare DNA zu entfernen, und führen Sie dann RCA durch, um Kreise zu amplifizieren; erstellen Sie eine Illumina-Bibliothek.
- Erwartete Ausgabe: Kreisaufrufe mit Junction-Koordinaten; erhöhte Abdeckung über EGFR; Split-Read-Unterstützung über Junctions hinweg.
- Validierung: Nach außen gerichtete PCR über die Junction und Sanger-Sequenzierung; optionale FISH zur Lokalisierung.
- Hinweise: RCA-Bedingungen können zu kleinen Kreisen tendieren; ziehen Sie eine ergänzende Tn5-basierte Detektion von Junctions in Betracht und, falls die Ressourcen es erlauben, eine Langlesevalidierung für größere Strukturen.
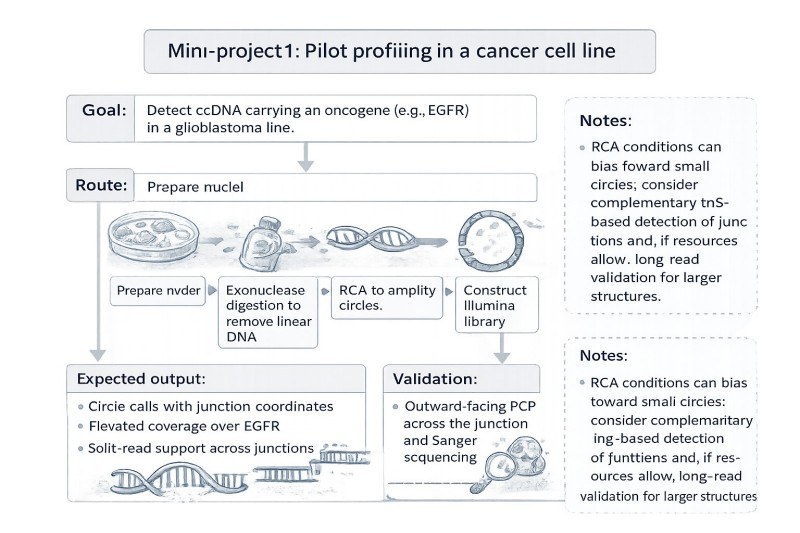 Abbildung 3. Pilot-Workflow für die ecDNA-Profilierung in einer Krebszelllinie.
Abbildung 3. Pilot-Workflow für die ecDNA-Profilierung in einer Krebszelllinie.
Mini-Projekt 2: Erkennung eines Kreisübergangs aus gesplitteten Reads (ATAC-seq)
- Ziel: Identifizierung von Prä-Amplifikationsjunctionen in derselben Linie unter Verwendung von ATAC-seq-Signaturen.
- Route: Führen Sie ATAC-seq durch; analysieren Sie auf Split-Reads und nach außen gerichtete Paare, die nicht linear kartiert sind; kennzeichnen Sie Kandidaten in der Nähe von EGFR.
- Erwartete Ergebnisse: Junction-Kandidaten, die durch inverse PCR bestätigt wurden; potenzielle Hotspots, an denen ecDNA später auftaucht.
- Hinweise: ATAC-seq erfasst zugängliche Chromatin; Tiefe und bioinformatische Filter sind wichtig. Siehe methodologischen Kontext in Kumar (2020).
Fehlerbehebungskasten: häufige Fallstricke und Kontrollen
- mtDNA-Übertragung: Da mtDNA zirkulär ist, kann unvollständige Reinigung die "Kreis"-Signale erhöhen. Fügen Sie eine mtDNA-spezifische Quantifizierung als Hintergrundüberprüfung hinzu.
- Unvollständige Exonuklease-Digestion: Restliches lineares DNA kann Junction-Signaturen nachahmen. Verwenden Sie Spike-in-Plasmid-Kontrollen, um die Verhältnisse von zirkulär zu linear zu quantifizieren und die Verdauung zu optimieren.
- RCA-Concatenar-Artefakte: Überverstärkung kann künstliche Chimären erzeugen. Titration von phi29, Begrenzung der Reaktionszeit und Einbeziehung einer Nach-RCA-Reinigung. Ziehen Sie eine Nicht-RCA-Anreicherung für kleine Zirkeln in Betracht, wenn Artefakte dominieren.
- Einschränkungs-/Fragmentierungsbias: Enzyme wie MspI legen Motivpräferenzen fest. Wenn Ihre Studie unterschiedliche Motivinhalte umfasst, ziehen Sie ergänzende Methoden in Betracht.
- Unzureichende Sequenzierungstiefe/-länge: Geringe Tiefe verpasst Junctions; fügen Sie lange Reads oder inverse PCR für wichtige Ziele hinzu.
Bioinformatik-Überblick: von Reads zu Kreisen
Anfänger fragen oft: „Wie werden die Ergebnisse aussehen?“ Hier ist eine kurze Übersicht über die erwarteten Ergebnisse und gängige Werkzeuge.
Erkennungslogik (Kurzlese)
- Geteilte Reads: Einzelne Reads, die sich an zwei benachbarte genomische Segmente anlagern, die im Referenzgenom nicht zusammenhängend sind und die Kreisgrenze überschreiten.
- Diskordante Paare: Mate-Paar-Orientierungen und Abstände, die auf eine geschlossene Fusion hindeuten.
- Junctionsunterstützung: Werkzeuge zählen unterstützende Reads und schätzen das Vertrauen; nach außen gerichtete PCR bietet orthogonale Validierung.
Werkzeuge, auf die Sie stoßen werden
- Circle-Map und verwandte Short-Read-Tools berichten über Kandidatenkreise mit Koordinaten, Längen und Leseunterstützungsmetriken. Siehe eine integrierte Diskussion in Fang (2024) über eccDNA-pipe und Werkzeugvergleiche.
- AmpliconArchitect (AA) rekonstruiert amplifizierte Strukturen, die häufig für ecDNA-Amplikons bei Krebs verwendet werden.
- Langzeit-Workflows (z. B. CReSIL) helfen dabei, große Kreise zu durchlaufen und interne Umstellungen abzubilden. Siehe vergleichende Anmerkungen in Gao (2024).
Erwartete Berichtsfelder (minimale Vorlage)
- Kreis-ID und genomische Koordinaten (z. B. hg38 chr7:54.000.000–54.250.000).
- Junction-Leseunterstützung (geteilte Reads, diskordante Paare) und Vertrauensscore.
- Länge und Sequenzzusammensetzung (Wiederholungen, Enhancer, Genüberlappung).
- Beispielmetadaten (Zelllinie, Behandlung) und Bedingung.
- Kopienzahlenschätzung oder abdeckungsbasierte Häufigkeitsmetriken.
Berichtswesen und Qualitätsstandards (Anfänger-Checkliste)
- Anreicherungseffizienz: Dokumentieren Sie die zirkulären:linearen Verhältnisse unter Verwendung von Spike-in-Plasmiden oder endogenen Referenzen (mtDNA), mit dem Ziel, hohe Post-Anreicherungsverhältnisse zu gewährleisten, um die Spezifität sicherzustellen.
- Hintergrund/chimäre Raten: Verfolgen Sie potenzielle RCA-Konkatenationsartefakte und Junction-Aufrufe mit geringer Unterstützung; ziehen Sie orthogonale Validierung in Betracht.
- Reproduzierbarkeit: Berichten Sie über die Übereinstimmung der Wiederholungen und die pro Probe normalisierten eccDNA-Zählungen in Bezug auf die Sequenzierungstiefe.
- Validierung: Einschluss einer externen PCR/Sanger-Bestätigung für repräsentative Junctions; optionale FISH für die Lokalisierung großer ecDNA.
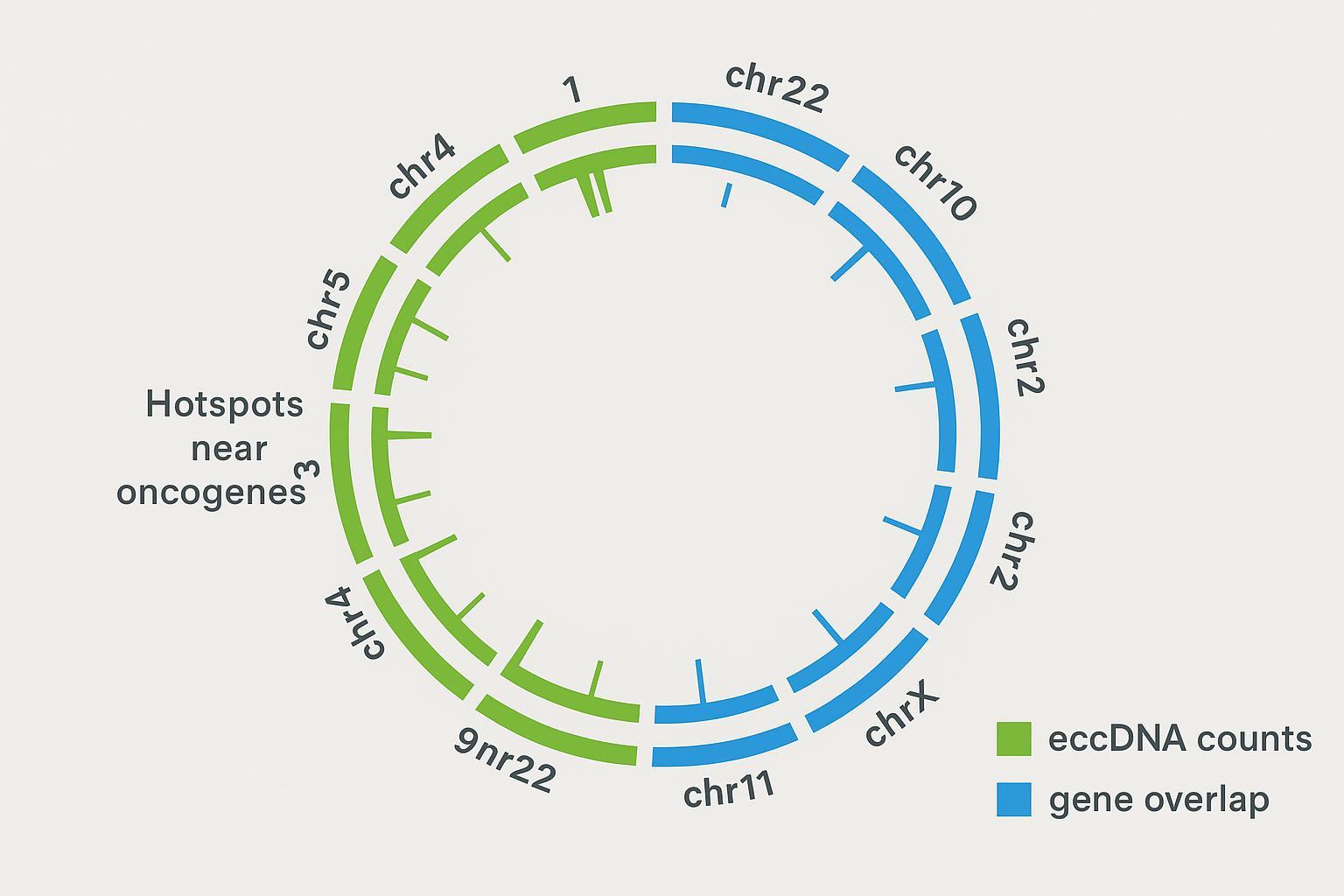 Abbildung 4: Beispiel im Circos-Stil mit Markenfarben. Der grüne Track zeigt hypothetische eccDNA-Zahlen an; blaue Markierungen kennzeichnen Genüberlappungen. Hotspots in der Nähe bekannter Onkogene und repetitiver Regionen sind zur Orientierung beschriftet.
Abbildung 4: Beispiel im Circos-Stil mit Markenfarben. Der grüne Track zeigt hypothetische eccDNA-Zahlen an; blaue Markierungen kennzeichnen Genüberlappungen. Hotspots in der Nähe bekannter Onkogene und repetitiver Regionen sind zur Orientierung beschriftet.
Optionale nächste Lektüre in der Reihe: ein tieferer Einblick in Pipelines, das Filtern von Artefakten und Berichtsstandards in Bioinformatik für eccDNA: Erkennung, Filterung und Berichterstattung.
Wenn Sie planen, die Analyse auszulagern, fordern Sie ein Ergebnis-Paket an, das Schnittpunktkoordinaten, Leseunterstützungsmetriken und Genannotationen umfasst – viele Teams arbeiten mit CD Genomics zusammen für NGS-Bioinformatikanalyse um veröffentlichungsreife Berichte zu erstellen.
Studienentwurf Überlegungen: Probenarten, Tiefe und Kontrollen (RUO)
Die Planung einer Studie, die interpretierbare eccDNA-Daten generiert, reduziert sich auf drei praktische Entscheidungen: welche Proben Sie analysieren, wie tief Sie sequenzieren und welche Kontrollen Sie einbeziehen.
Probenarten und -entnahme
- Kultivierte Zellen: Bieten saubere Kerne für exonukleasebasierte Arbeitsabläufe; leicht skalierbar und reproduzierbar.
- Tumorgewebe: Heterogenität erschwert die Interpretation; ziehen Sie Mikrodissektion oder Einzelzell-Anpassungen in Betracht, wenn möglich.
- Plasma/cfDNA: Nicht-invasive Probenentnahme; erwarten Sie ausgeprägte Spitzen in der Kreisdurchmessergröße und einen höheren Hintergrund; enzymatische Reinigung ist entscheidend.
Tiefe und Plattformwahl
- Short-Read (Illumina): Häufig für Circle-Seq; ziele auf eine ausreichende Tiefe, um niedrig-abundante Kreise zu erkennen und robuste Junction-Calls zu unterstützen.
- Langzeitlesung (ONT/PacBio): Erhöht das Vertrauen in große Zyklen und komplexe Strukturen; planen Sie, wenn möglich, eine Abdeckung von >10X bei den interessierenden Zielen.
- ATAC-seq Ergänzung: Nützlich, um Junctions und zugängliche Regionen zu kennzeichnen, die der ecDNA-Amplifikation vorausgehen können.
Kontrollen und Qualitätssicherung
- Spike-ins: Plasmid-Spike-ins zur Schätzung der zirkulären Anreicherungs-effizienz und zur Überwachung von Verlusten.
- Negative Controls: Mock-Digestionsproben zur Aufdeckung unvollständiger Entfernung von linearem DNA; No-Template-Kontrollen zur Erkennung von Amplifikationsartefakten.
- Orthogonale Validierung: Auswärtige PCR und Sanger; optionale FISH oder Pulsfeld-Gelelektrophorese (PFGE) für Reinheitsprüfungen.
Für QC-Metriken in eccDNA-Projekten – Anreicherungs-effizienz, Hintergrund, Reproduzierbarkeit – siehe den Serienleitfaden zu Qualitätsmetriken für eccDNA-Sequenzierung.
FAQ für Erstleser von eccDNA
- Bedeuten eccDNAs und ecDNA dasselbe? Nein. "eccDNA" ist der Überbegriff für verschiedene zirkuläre DNA-Moleküle. "ecDNA" bezieht sich normalerweise auf große, amplifikationstragende Zirkeln, die häufig in Krebs vorkommen und oft ganze Onkogene tragen. Für Größen-/Funktionsunterscheidungen siehe Wang (2024) und Zhou (2024).
- Sind kleine eccDNAs nur Artefakte der Bibliotheksvorbereitung? Nicht im Allgemeinen. Mehrere Assays – einschließlich nicht-RCA-Anreicherungen und ATAC-seq-Junktionsdetektion – unterstützen die Existenz kleiner eccDNAs in verschiedenen Kontexten. Die Wahl der Methode und Kontrollen sind wichtig, um RCA-Concatemere oder unvollständige lineare DNA-Aufreinigungen zu vermeiden. Siehe methodologische Diskussionen in Yu (2023) und Kumar (2020).
- Welche Proben können profiliert werden? Viele: kultivierte Zellen, Gewebe und sogar Plasma/cfDNA. Plasma-Studien, die Exonuklease V (ExoV) und Restriktionsenzyme (z. B. MspI) verwenden, haben unterschiedliche Größenpeaks für eccDNA im Vergleich zu linearer cfDNA berichtet, was die nucleosomale Periodizität widerspiegelt. Siehe Sin (2020) in PNAS.
- Wie validiere ich Kreisaufrufe? Verwenden Sie nach außen gerichtete PCR über Übergänge und Sanger-Sequenzierung; FISH kann große ecDNA lokalisieren; Langzeit-Sequenzierung oder inverse PCR können zusätzliche Bestätigungen liefern.
- Gibt es eine Datenbank bekannter Kreise? Aggregatoren wie CircleBase V2 und eccDNABase sammeln Koordinaten, Annotationen und Probenmetadaten über verschiedene Arten. Siehe CircleBase V2 (2025) und eccDNABase (2024).
Fazit und nächste Schritte (RUO)
EccDNA-Sequenzierung erweitert unsere Sicht auf die Struktur und Variabilität des Genoms. Durch die Messung von Junctions, Geninhalt und Amplifikation über die Chromosomen hinaus können Forscher verfolgen, wie Kreise die Heterogenität von Krebs beeinflussen und ihre Rollen in der somatischen Biologie und im Altern erkunden. Der Schwung des Feldes – sichtbar in den Publikationen "eccdna nature" – resultiert aus der Verbindung klarer Beweise (Junctions, Geninhalt, Kopienzahl) mit bedeutungsvollen biologischen Fragen.
Wenn Sie ein RUO-Projekt planen und externe Unterstützung benötigen, bieten Anbieter wie CD Genomics relevante Dienstleistungen an. Erkunden Sie Lösungen für die Onkologie-Forschung. bei CD Genomics und umfassend Bioinformatik-Dienstleistungen um das Studiendesign und die Berichtserfordernisse zu besprechen.
Für ein tieferes Lernen in dieser Reihe:
- Methoden und Kontrollen: Auswahl von eccDNA-Anreicherungsmethoden
- End-to-End Laboranleitung: Experimenteller Arbeitsablauf für die Sequenzierung von eccDNA
- Krebsanwendungen: eccDNA bei Krebs
- Altern und somatische Biologie: eccDNA in somatischen Zellen und Alterung
- Bioinformatik und Berichterstattung: Bioinformatik für eccDNA: Erkennung, Filterung und Berichterstattung
- QC und Reproduzierbarkeit: Qualitätsmetriken für eccDNA-Sequenzierung
Für die Unterstützung des RUO-Projekts – Studiendesign, Sequenzierung und nachgelagerte Berichterstattung – kontaktieren Sie CD Genomics. Onkologie-Forschungslösungen und Lösungen zur Analyse genomischer Daten den experimentellen Umfang und die Ergebnisse zu besprechen.
Referenzen:
- Wang, X. et al., "Die Geheimnisse der extrachromosomalen zirkulären DNA enthüllen" (Überblick, 2024) — autoritativer Open-Access-Überblick, der die Klassen, Größen und Funktionen von eccDNA zusammenfasst; siehe Wang 2024 Bewertung (PMC).
- Zhou, Y., "ecDNA-Merkmale und Tumorverteilung" (Frontiers in Genetics, 2024) — Diskussion über ecDNA-Definitionen und Tumorhäufigkeit; siehe Zhou 2024, Frontiers in Genetics.
- Zeng, W. et al., "Landschaft von eccDNA in der normalen Hämatopoese und der Evolution von Leukämie" (Genome Research, 2022) — Open-Access-Landschaftspapier zur Verteilung von eccDNA in verschiedenen Geweben; siehe Zeng 2022, Genomforschung (PMC).
- Zhao, X. et al., "Rollen und Tumorassoziation von extrachromosomaler zirkulärer DNA" (eLife, 2022) — Übersicht über die Biologie von eccDNA und ihre Verbindungen zu Krankheiten; siehe Zhao 2022, eLife.
- Kumar, P. et al., "ATAC-seq identifiziert extrachromosomale zirkuläre DNA-Verbindungen" (Science Advances, 2020) — zeigt ATAC-seq-Signaturen zur Erkennung und Validierung von eccDNA-Verbindungen; siehe Kumar 2020, Science Advances und der Open-Access-Datensatz Kumar et al. (PMC).
- Yu, L. et al., "Circle-Seq-Varianten und praktische Einblicke" (Methodenübersicht, 2023) — praktische Anmerkungen zur Synthese und Validierung von Circle-Seq; siehe Yu 2023 (PMC).
- Møller, H.D., "Circle-Seq-Protokoll" (Nature Protocols / PubMed, 2020) — ursprüngliches Circle-Seq-Protokoll und schrittweise Anleitung; siehe Møller 2020 Protokoll (PubMed).
- Fang, Z. et al., "eccDNA-pipe und Werkzeugvergleiche" (Briefings in Bioinformatics, 2024) — integrierter Workflow und Werkzeugbenchmarking zur eccDNA-Erkennung; siehe Fang 2024, Briefings in Bioinformatik.
- Gao, Y. et al., "Langzeit-Leserekonstitution und Anreicherungsabgleich (CReSIL)" (Methodenvergleich, 2024) — Diskussion über die Leistung der Langzeit-Leserekonstitution und Anreicherung; siehe Gao 2024 (PMC).
- Sin, H.S. et al., "Plasma eccDNA-Anreicherung mit ExoV und Restriktionsenzymen" (PNAS, 2020) — Methoden und Größenprofilbeobachtungen für Plasma-eccDNA; siehe Sin 2020, PNAS.
- National Cancer Institute, Krebsgenomik-Programm — "Überblick über ecDNA und Implikationen" (NCI CCG Blog, 2022) — zugängliche Zusammenfassung der ecDNA-Biologie bei Krebs; siehe NCI CCG ecDNA Übersicht (2022).
- CircleBase V2 — kuratierte eccDNA-Datenbank (2025 Landing/PMC-Artikel) — aggregierte Koordinaten und Annotationen über Studien hinweg; siehe CircleBase V2 (PMC).
- eccDNABase — umfassende Beschreibung und Ressource der eccDNA-Datenbank (MBE/OUP) — siehe eccDNABase (2024/2025, MBE).
Autor
Yang H. — Senior Scientist, CD Genomics; Universität Florida.
Yang ist ein Genomforschungswissenschaftler mit über 10 Jahren Forschungserfahrung in Genetik, molekularer und zellulärer Biologie, Sequenzierungsabläufen und bioinformatischer Analyse. Er ist sowohl in Labortechniken als auch in der Dateninterpretation versiert und unterstützt das Design von RUO-Studien und NGS-basierten Projekten.


