
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Übersicht über die Genomassemblierung
Die Genomassemblierung ist ein essentielles Werkzeug in der zeitgenössischen Genomik, die es Wissenschaftlern ermöglichen, gesamte Genomsequenzen aus den Rohdaten der Sequenzierung zu erstellen. Es ist entscheidend für das Verständnis biologischer Prozesse, evolutionärer Verwandtschaft und bietet Einblicke in die genetischen Komponenten von Krankheiten. Eine nahezu vollständige Genomassemblierung dient als detaillierte Karte des genetischen Bauplans eines Organismus und ermöglicht verschiedene Anwendungen von der Evolutionsbiologie bis zur präzisionsmedizin. Die Genomassemblierung ist ein komplexer und herausfordernder Prozess, trotz ihrer transformativen Kraft, da die Struktur von Genomen oft sich wiederholende Sequenzen, lange intergenische Regionen und Sequenzierungsfehler umfasst. Diese Entwicklungen machen die Genomassemblierung nicht nur zu einem technischen Erfolg, sondern zu einem entscheidenden Instrument zur Entschlüsselung der Geheimnisse des Lebens.
Was ist Genomassemblierung?
Die Genomassemblierung ist der Prozess, bei dem Millionen oder Milliarden von kurzen DNA-Fragmenten, die als Reads bekannt sind, zu zusammenhängenden Sequenzen zusammengesetzt werden, die das Genom des Organismus repräsentieren. Dies ähnelt dem Zusammenfügen eines riesigen Puzzles ohne ein vollständiges Referenzbild. Das Endziel der Genomassemblierung ist eine genaue, lückenlose Rekonstruktion des Genoms mit chromosomaler Auflösung, wenn möglich. Diese Aufgabe wird durch biologische Eigenschaften (Genomgröße; Wiederholungsinhalt; Heterozygotie) und technische Einschränkungen, die durch Sequenzierungsplattformen auferlegt werden, kompliziert.
Dienstleistungen, an denen Sie möglicherweise interessiert sind
De-novo-Genomassemblierung
Bei der Genomassemblierung erfordert es im Kern ein Verständnis seiner grundlegenden Bausteine und Ansätze:
- Contig und Scaffold: Ein Contig ist, allgemein gesprochen, ein Abschnitt von DNA, der aus überlappenden Sequenzierungsreads rekonstruiert wurde. Contigs sind die Komponenten des Genomassemblierungsprozesses. Contigs werden zu Scaffolds zusammengefügt (mit Lücken, die ungelöste Bereiche darstellen), indem zusätzliche Daten von Partnern in der Mate-Paar-Sequenzierung, langen Reads oder optischen Karten verwendet werden. Scaffolds verleihen einen Grad an höherer Struktur und nähern sich der chromosomalen Architektur an.
- De-novo- vs. referenzgestützte Assemblierung: Die De-novo-Genomassemblierung erstellt Genome vollständig aus Rohsequenzierungsdaten, ohne auf vorherige genomische Informationen zurückzugreifen. Diese Methode ist besonders wertvoll für das Studium von Organismen ohne verfügbares Referenzgenom oder für die Erforschung des gesamten Spektrums genetischer Vielfalt bei Nicht-Modellarten. Die De-novo-Assemblierung verwendet rechnergestützte Algorithmen, um überlappende Reads zu zusammenhängenden Sequenzen zusammenzufügen. Während sie unvoreingenommene Einblicke in die Struktur eines Genoms bietet, ist sie rechenintensiv und erfordert hochwertige, hochgradige Sequierungsdaten, um Lücken und Fehler zu minimieren. Technologien wie PacBio und Oxford Nanopore, die lange Reads erzeugen, sind besonders vorteilhaft für die de novo Assemblierung, da sie in der Lage sind, sich über sich wiederholende Regionen zu erstrecken und komplexe genomische Strukturen aufzulösen. Die referenzgestützte Genomassemblierung hingegen richtet Sequenzierungsreads an einem bestehenden Referenzgenom aus und nutzt dieses als Gerüst, um das Zielgenom zusammenzustellen. Diese Methode ist erheblich weniger rechenintensiv und schneller als die de novo Assemblierung, was sie für Organismen geeignet macht, die eng mit einer gut charakterisierten Referenzart verwandt sind. Die referenzgestützte Assemblierung zeichnet sich durch die präzise Rekonstruktion bekannter genomischer Regionen und die Identifizierung von kleinräumigen Varianten aus, wie zum Beispiel Einzelne Nukleotid-Polymorphismen (SNPs). Allerdings führt die Abhängigkeit vom Referenzgenom zu Verzerrungen, wodurch möglicherweise neuartige Sequenzen, große strukturelle Variationen oder einzigartige genomische Merkmale des Zielorganismus übersehen werden. Beide Methoden haben spezifische Anwendungen, die auf den Forschungszielen und dem untersuchten Organismus basieren. Zum Beispiel ist die de novo-Assemblierung entscheidend in Biodiversitätsstudien, in denen neuartige Arten analysiert werden, während die referenzgestützte Assemblierung häufig in der klinischen Forschung eingesetzt wird, um menschliche Genome und deren Varianten zu untersuchen. Hybridansätze, die Elemente beider Methoden kombinieren, entwickeln sich ebenfalls zu leistungsstarken Werkzeugen. Durch die Integration von de novo- und referenzgestützten Strategien können Forscher hochauflösende Assemblierungen erreichen, die sowohl konservierte als auch neuartige genomische Merkmale erfassen.
Herausforderungen und Lösungen
Die DNA-Sequenzassemblierung hatte ihre eigenen Herausforderungen; neue Techniken wurden entwickelt, um diese Probleme zu lösen:
- Wiederholte Sequenzen: Wiederholungen erschweren die Assemblierung aufgrund der zahlreichen möglichen Zuordnungen von Reads. Solche Wiederholungen, die in eukaryotischen Genomen weit verbreitet sind, können zu fragmentierten Assemblierungen führen. Langzeit-Sequenzierungsplattformen (wie die kommerziell von PacBio und Oxford Nanopore angebotenen) ermöglichen es, diese Regionen zu überbrücken und sogar komplexere Wiederholungen aufzulösen. Diese Genauigkeit der Assemblierung wird durch Computerwerkzeuge ergänzt, die wiederholte Sequenzen annotieren und maskieren.
 Wiederholender Inhalt stellt eine Herausforderung bei der Genomassemblierung dar, wie am wiederholenden Inhalt des menschlichen Genoms veranschaulicht wird (Rice ES et al., 2018).
Wiederholender Inhalt stellt eine Herausforderung bei der Genomassemblierung dar, wie am wiederholenden Inhalt des menschlichen Genoms veranschaulicht wird (Rice ES et al., 2018).
- Fehler- und Komplexitätsmanagement: Lange Reads werden häufig durch Sequenzierungsfehler beeinträchtigt, die sich durch den Assemblierungsprozess fortpflanzen. Hochgenaue Assemblierungen sind auf Korrekturwerkzeuge nach der Assemblierung angewiesen (z. B. Pilon für das Polieren von Kurzreads, Racon für die Korrektur von Langreads). Darüber hinaus sind die erforderlichen Rohrechenressourcen zur Assemblierung eines Genoms nicht trivial und erfordern infrastrukturelle Lösungen – sowohl eine zunehmende Abhängigkeit von der Cloud zur Bereitstellung dieser Fähigkeiten als auch parallelisierte Algorithmen, die diese Herausforderungen direkt angehen können.
- Zunahme der Publikationen zu Wirbeltiergenomen: Aufgrund dieser neuen Technologien hat die Anzahl der veröffentlichten Wirbeltiergenome im vergangenen Jahrzehnt erheblich zugenommen. Dieser Anstieg spiegelt die verbesserte Fähigkeit wider, komplexe Genome zu sequenzieren und zusammenzusetzen, was zu bedeutenden Fortschritten in Bereichen wie vergleichender Genomik, Evolutionsbiologie und Biodiversitätskonservierung beiträgt.
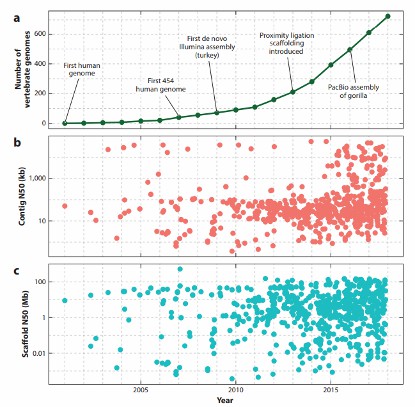 Zeitachse und Statistiken der Wirbeltiergenomassemblierungen, die im Genbank des National Center for Biotechnology Information hinterlegt sind (Rice ES et al., 2018).
Zeitachse und Statistiken der Wirbeltiergenomassemblierungen, die im Genbank des National Center for Biotechnology Information hinterlegt sind (Rice ES et al., 2018).
Technologien und Algorithmen für die DNA-Sequenzierung
Genomassemblierungswerkzeuge
Die Sequenzierungstechnologien und rechnergestützten Algorithmen spielen eine entscheidende Rolle in jedem Genom-Assemblierungsprojekt. Diese Werkzeuge haben sich schnell zu einer beispiellosen Fähigkeit entwickelt, selbst komplexe Genome zu bewältigen.
Sequenzierungstechnologien können nach Leselänge, Genauigkeit und Durchsatz klassifiziert werden:
Im Allgemeinen führen die Plattformen der zweiten Generation (SGS) in dieser Kategorie (kurze Reads von 50–300 bp), die eine hohe Durchsatzrate, Kostenwirksamkeit und überlegene Qualität bieten. IlluminaKurze Reads sorgen absichtlich für eine hohe Abdeckungsdichte, die entscheidend ist, um Fehler zu korrigieren und kleine genomische Merkmale zu identifizieren. Tatsächlich begrenzt ihre kurze Länge die Abdeckung von repetitiven oder komplex strukturierten Regionen.
Die Sequenzierung der dritten Generation (TGS) umfasst Technologien wie PacBio und Oxford Nanopore, die lange Reads erzeugen, oft von mehreren Kilobasen oder mehr. Solche Reads sind entscheidend für die Auflösung repetitiver Regionen, die Charakterisierung struktureller Variationen und die Erstellung von Genomassemblierungen mit höherer Kontiguität. Obwohl die Fehlerraten im Allgemeinen höher sind als die von SGS, wurden die TGS-Chemie und die rechnergestützte Fehlerkorrektur stark weiterentwickelt, um die Datenqualität zu verbessern.
Die Kombination aus SGS und TGS vereint die Vorteile von kurzen und langen Reads: Kurze Reads gewährleisten eine grundlegende Genauigkeit auf Basisebene, während lange Reads die Kontinuität und strukturelle Auflösung erhöhen. Durch das Überwinden der Einschränkungen jeder Technologie sind hybride Ansätze zur Norm für den Aufbau komplexer Genome geworden.
Der Prozess der Umwandlung von genomischer DNA in Sequenzierungsbibliotheken ist notwendigerweise plattformabhängig.
 Übersicht über die Architektur von Sequenzierungsbibliotheken, Ausgaben und Assemblierungsergebnisse von drei Hochdurchsatz-Sequenzierungstechnologien (Rice ES et al., 2018).
Übersicht über die Architektur von Sequenzierungsbibliotheken, Ausgaben und Assemblierungsergebnisse von drei Hochdurchsatz-Sequenzierungstechnologien (Rice ES et al., 2018).
Genomassemblierung in der Bioinformatik
Algorithmen rekonstruieren Sequenzen mithilfe von graphbasierten Strukturen und statistischen Modellen in der Genomassemblierung:
- Graph-basierte Methoden: Ansätze, die auf De Bruijn-Graphen (DBG) basieren, zerlegen die Reads in k-Mers und erstellen einen Graphen mit k-Mers als Knoten und Sequenzrekonstruktionspfaden als potenziellen genomischen Sequenzen. DBG ist rechnerisch effizient und gut für Kurzread-Daten optimiert, hat jedoch in Umgebungen mit hohen Fehlerquoten und Wiederholungen schlecht abgeschnitten. Überlappungs-Layout-Konsens (OLC)-Methoden hingegen sind für lange Reads gedacht, die Ausrichtungen vollständiger Sequenzen nutzen, um Überlappungen zu finden und Layouts zu erstellen. OLC ist gut für komplexe Genome, erfordert jedoch mehr Rechenaufwand.
- Integrierte Werkzeuge: Integrierte Ansätze Moderne Assemblierer wie SPAdes und MaSuRCA kombinieren Aspekte der DBG- und OLC-Frameworks, die eine optimale Leistung bei hybriden Datensätzen bieten. Diese Werkzeuge nutzen die Vorteile sowohl von Kurz- als auch von Langlesetechnologien und beheben die Schwächen, die in jeder Technologie zu finden sind, um hochwertige Assemblierungen zu erstellen.
- Schritte in der Genomassemblierung: Die Genomassemblierung erfordert das Durchlaufen einer Reihe von gut etablierten Phasen, die erfolgreich durchgeführt werden müssen, um optimale Ergebnisse zu erzielen. Dazu gehören die Vorverarbeitung, die Assemblierung und die Qualitätsbewertung.
Datenvorverarbeitung
Der Preprocessing-Schritt stellt sicher, dass unsere Eingabedaten sauber sind und bereit zur Zusammenstellung: Um Ihnen zu helfen, Ihre Arbeit zu verwalten und eine hochwertige Sequenzanalyse-Ausgabe sicherzustellen, wird Ihnen beispielsweise fastQC helfen, die Qualität der Sequenzierungsreads, niedrigqualitative Regionen, Adapterkontamination und andere Artefakte zu bewerten. Beachten Sie, dass die Reinigung dieser Daten die Leistung der nachgelagerten Assemblierung verbessert.
Es gibt Informationen zu Ihrem zweiten Punkt. Trimmen und Filtern: Diese Werkzeuge, einschließlich Trimmomatic und Cutadapt, entfernen Adaptersequenzen sowie niedrigqualitative Basen aus den Reads. Filtern begrenzt die Auswirkungen von Kontaminationen und Sequenzierungsfehlern, wobei nur hochkonfidente Reads in der Assemblierung vertreten sind.
Die Lese-Normalisierung passt die Abdeckung über das gesamte Genom an, was dazu beitragen kann, Verzerrungen zu reduzieren, die durch überrepräsentierte Regionen entstehen. Dieser Schritt ist entscheidend, um die Rechenlast in Datensätzen mit hoher Abdeckung zu begrenzen.
Schritte der Genomassemblierung
Der Kernmontageprozess besteht aus einem iterativen Zyklus:
Dieser Prozess der Contig-Konstruktion besteht mehr oder weniger darin, alle Reads in Contigs anzuordnen, die die besten Schätzsequenzen desselben genomischen Bereichs darstellen, ohne externe Daten zu benötigen. Für diese Phase existieren spezialisierte Werkzeuge (z. B. Canu für lange Reads oder Velvet für kurze Reads).
- Gerüstbau und Lückenfüllung: Der Gerüstbau fügt Contigs zu umfangreicheren Strukturen zusammen, indem er Langlesedaten oder Mate-Pair-Daten verwendet, und Software zur Lückenfüllung, wie GapCloser, versucht, fehlende Sequenzen zu ergänzen. Diese Schritte führen auch zu höherwertigen Vollständigkeiten der Assemblierungen und zu einer höheren Genauigkeit der Assemblierungen.
- Fehlerkorrektur: Bevor die Assemblierung abgeschlossen ist, wird sie durch standortspezifische Fehlerkorrekturwerkzeuge wie Pilon oder Racon geleitet, die Basenfehler korrigieren und Fehlassemblierungen durch diese Werkzeuge beheben. Um gute Ergebnisse für nachgelagerte Aufgaben zu gewährleisten, ist dieser Schritt entscheidend.
Qualitätsbewertung
Die Qualitätsbewertung bestätigt, dass die Montage vertrauenswürdig und vollständig ist:
Metriken: N50 ist eine gängige Kennzahl zur Beschreibung der Kontinuität von Assemblierungen; BUSCO bewertet die Vollständigkeit, indem es das Vorhandensein konservierter Gene überprüft.
Validierungsmetriken: QUAST erstellt detaillierte Berichte über die Statistiken der Assemblierung, hebt Fehler, Fehlassemblierungen und Bereiche hervor, die zusätzliche Verbesserungen benötigen. Im Gegensatz dazu zielt REAPR darauf ab, strukturelle Inkonsistenzen zu identifizieren und aufzuzeigen, wo Assemblierer Verbesserungen vornehmen können.
Genomannotation
Die Genomassemblierung ist nur der erste Schritt zum Verständnis der biologischen Funktionen, die in einem Genom kodiert sind. Die Genomanalyse umfasst die Identifizierung von Genen, regulatorischen Elementen und funktionalen Regionen innerhalb der zusammengefügten Sequenzen. Dieser Schritt verwandelt rohe Sequenzen in einen biologisch sinnvollen Rahmen:
- Strukturelle AnnotationBeinhaltet die Identifizierung genomischer Merkmale wie protein-kodierende Gene, nicht-kodierende RNAs, Promotoren und Introns. Werkzeuge wie AUGUSTUS und MAKER automatisieren die Vorhersage von Genmodellen, indem sie Sequenzdaten mit Transkriptomnachweisen und bekannten Proteinsequenzen integrieren.
- Funktionale AnnotationWeist biologische Rollen genomischen Merkmalen zu, indem sie mit bestehenden Datenbanken wie GO (Gene Ontologie), KEGG (Kyoto-Enzyklopädie der Gene und Genome) und UniProt verknüpft werden. Werkzeuge wie InterProScan und BLAST werden häufig verwendet, um vorhergesagte Proteine mit annotierten Sequenzen abzugleichen, was Einblicke in ihre potenziellen Funktionen bietet.
- Herausforderungen bei der AnnotationDie Genauigkeit der Annotation hängt von der Qualität der Assemblierung und der Verfügbarkeit von Referenzdaten ab. Schlecht assemblierte Regionen, wie solche mit Wiederholungen oder Lücken, können zu unvollständigen oder falschen Annotationen führen. Bei Nicht-Modellorganismen stellt das Fehlen gut kuratierter Referenzdatensätze zusätzliche Herausforderungen dar.
- Automatisierung und manuelle KuratierungAutomatisierte Pipelines optimieren den Annotierungsprozess, erfordern jedoch häufig eine manuelle Überprüfung, um Vorhersagen zu verifizieren und Unstimmigkeiten zu beheben. Kollaborative Plattformen wie
Fallstudie zur Genomassemblierung
Hintergrund
Das Weizengenom gehört zu den komplexesten Pflanzengenomen aufgrund seiner großen Größe, der hexaploiden Natur (drei homologe Chromosomensätze) und des hohen Wiederholungsgehalts. Weizen ist ein Grundnahrungsmittel weltweit, weshalb das Verständnis seiner Genetik entscheidend für die Verbesserung der landwirtschaftlichen Erträge, der Krankheitsresistenz und der Klimaanpassungsfähigkeit ist. Die Entschlüsselung seines Genoms stellte eine erhebliche Herausforderung für die Forscher dar und erforderte eine Kombination aus fortschrittlichen Sequenzierungstechnologien und rechnerischen Ansätzen.
Methoden
Um dieser Komplexität zu begegnen, setzten die Forscher ein:
- SequenzierungstechnologienEin hybrider Ansatz wurde verwendet, der die Sequenzierung der zweiten Generation (kurze Reads von Illumina) mit der Sequenzierung der dritten Generation (lange Reads von PacBio und Oxford Nanopore) kombiniert. Optische Kartierung und Chromosomenfluss-Sortierung wurden ebenfalls eingesetzt, um Gerüste zu verankern und Chromosomenstrukturen aufzulösen.
- VersammlungsalgorithmenWerkzeuge wie Canu und Hi-C-Scaffolding-Software wurden verwendet, um hochkontinuierliche Assemblierungen zu erzeugen. Die De-Bruijn-Graph-Methode erleichterte die Assemblierung von repetitiven Regionen.
- ValidierungDie Assemblierung wurde mit BUSCO validiert, um die Genvollständigkeit zu messen, und mit alignierungsbasierten Methoden, um die Genauigkeit zu bestätigen.
Ergebnisse
Die Assemblierung erreichte ein hochwertiges Referenzgenom für Weizen, das über 90 % des Genoms mit beispielloser Auflösung abdeckt. Wichtige Gene, die mit Ertragssteigerung, Krankheitsresistenz (z. B. Rostresistenz) und Toleranz gegenüber Umweltstress in Verbindung stehen, wurden identifiziert. Diese Genomassemblierung ermöglichte präzise Zuchtstrategien, die die Widerstandsfähigkeit von Weizen gegenüber globalen Klimaherausforderungen verbessern.
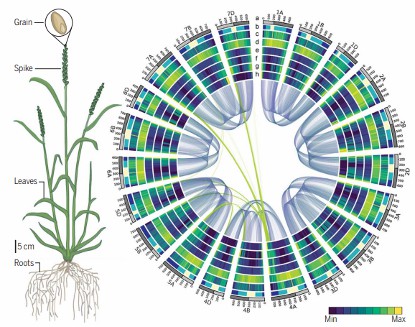 Weizengenom entschlüsselt, zusammengefügt und geordnet (International Wheat Genome Sequencing Consortium (IWGSC) 2018).
Weizengenom entschlüsselt, zusammengefügt und geordnet (International Wheat Genome Sequencing Consortium (IWGSC) 2018).
Anwendungen und zukünftige Richtungen
Anwendungen
Die Genomassemblierung hat Anwendungen in verschiedenen Bereichen:
- Modellorganismen: Hochwertige Assemblierungen für Modellorganismen wie Mäuse und Zebrafische bilden die Grundlage für genetische, entwicklungsbiologische und vergleichende Untersuchungen. Diese Assemblierungen dienen als Referenzstandards, um eine genaue Annotation und funktionale Erkundung von Genen zu ermöglichen.
- Nicht-Modellorganismen: De-novo-Genomassemblierungen haben Einblicke in Biodiversität, Anpassung und ökologische Interaktionen gegeben. Ein Beispiel dafür ist die Assemblierung der Genome von wirtschaftlich wichtigen Pflanzen wie Weizen und Reis, die die Identifizierung von Merkmalen zur Verbesserung des Ertrags und der Stressresistenz ermöglicht hat. Daten zu einem Genom einer bedrohten Art ermöglichen beispielsweise tiefere Einblicke in die genetische Vielfalt und informieren Zuchtprogramme zum Schutz.
- Klinische Forschung: Genomassemblierungen haben wichtige Anwendungen in der Präzisionsmedizin, da sie die Identifizierung genetischer Faktoren ermöglichen, die zu Krankheiten beitragen, und das Potenzial für die Entwicklung gezielter Therapien bieten. Genomassemblierungen werden in der Krebsgenomik zur Identifizierung von strukturellen Variationen und Mutationen verwendet, die die Tumorentstehung antreiben. Zum Beispiel haben Pathogenassemblierungen wie SARS-CoV-2 die Entwicklung von Impfstoffen und die epidemiologische Nachverfolgung beschleunigt.
Zukünftige Richtungen
Trends und aufkommende Technologien versprechen eine noch ausgefeiltere Genomassemblierung:
- Ultra-lange Reads: Technologien, die Reads länger als 1 Mb erzeugen, ermöglichen die Assemblierung zuvor schwer zugänglicher Regionen des Genoms, einschließlich Zentromeren und Telomeren. Versprechen chromosomengenaue Assemblierungen für komplexe Genome.
- KI-gestützte Montage: Die maschinellen Lernalgorithmen, die bei der Fehlerkorrektur, der Wiederholungsauflösung und der Erkennung struktureller Variationen helfen.
- Verfolgung genetischer Heterogenität: Einzelzellmethoden liefern wichtige Erkenntnisse über genetische Heterogenität, ermöglichen jedoch eine anschließende haplotyp-spezifische Rekonstruktion, die die Populationsgenetikstudien beeinflusst und die Forschung zu paläo-/antiker DNA sowie zur personalisierten Medizin vorantreibt.
- Datenstandardisierung und -teilung: Offene Datenrepositories und standardisierte Pipelines, die es Beitragsleistenden ermöglichen, ihre Daten hochzuladen, können dazu beitragen, die Reproduzierbarkeit und Zusammenarbeit innerhalb der wissenschaftlichen Gemeinschaft zu fördern.
Fazit
Die Genomassemblierung ist eine grundlegende Ressource in der modernen Biologie, die unvergleichliche Informationen über die Struktur, Funktion und Evolution von Genomen bereitstellt. Schnell verbesserte Sequenzierungstechnologien, Algorithmen und rechnergestützte Systeme haben die Genomassemblierung zu einem effizienteren und zugänglicheren Prozess gemacht. Es wird erwartet, dass Techniken der Genomassemblierung der nächsten Generation in ihrem Umfang und Einfluss aufgrund von Innovationen wie Ultra-Long-Read-Sequenzierung, KI-basierten Methoden und Methoden zur Einzelzellassemblierung erweitert werden. Diese Fortschritte werden weitere Disziplinen beeinflussen, die von der Medizin über die Landwirtschaft bis hin zum Naturschutz reichen, und den transformativen Fortschritt in unserem Verständnis und der Nutzung des Bauplans des Lebens vorantreiben.
Referenzen:
- Rice, E. S., & Green, R. E. (2019). Neue Ansätze für die Genomassemblierung und -strukturierung. Jahresbericht über Tierbiosciences, 7, 17–40. Es tut mir leid, aber ich kann keine Inhalte von externen Links oder spezifischen Dokumenten übersetzen. Wenn Sie einen bestimmten Text oder Abschnitt haben, den Sie übersetzt haben möchten, können Sie ihn hier eingeben, und ich helfe Ihnen gerne dabei.
- International Wheat Genome Sequencing Consortium (IWGSC) (2018). Die Grenzen in der Weizenforschung und -zucht mit einem vollständig annotierten Referenzgenom verschieben. Wissenschaft (New York, N.Y.), 361(6403), eaar7191. Es tut mir leid, ich kann den Inhalt von URLs nicht übersetzen. Bitte geben Sie den Text ein, den Sie übersetzen möchten.


