
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Jenseits des Manhattan-Plots: Fortgeschrittene statistische Strenge und Multi-Omik-Integration in der genomweiten Analyse
Die genomweite Analyse ist leicht zu stark zu vereinfachen. Viele Artikel stellen sie weiterhin als Suche nach signifikanten Loci im gesamten Genom dar, gefolgt von einem Manhattan-Diagramm und einer kurzen Liste der wichtigsten SNPs. Diese Beschreibung ist vertraut, aber sie verpasst den schwierigsten Teil der Arbeit. Die eigentliche Herausforderung besteht nicht darin, einen Peak zu erzeugen. Die wahre Herausforderung besteht darin, zu entscheiden, ob dieser Peak statistischer Überprüfung standhält, ob er die Phänotyp-Biologie und nicht die Kohortenstruktur widerspiegelt und ob er in eine glaubwürdige mechanistische Hypothese übersetzt werden kann.
Diese Unterscheidung ist in großen Datensätzen aus der Ära 2026 noch wichtiger. Die Kohorten sind größer. Die Variantenanzahl ist höher. Die Populationsstruktur ist komplexer. Auch die Erwartungen an die Ergebnisse sind höher. Eine Liste signifikanter Marker reicht für die meisten ernsthaften Forschungsprogramme nicht mehr aus. Die Teams möchten wissen, ob die Analyse die falschen Entdeckungen gut kontrolliert hat, ob verborgene Verwandtschaft das Signal verzerrt hat, ob der führende SNP tatsächlich kausal ist und ob das Locus auf eine vertretbare Weise mit Expression, Spleißen oder regulatorischer Funktion verbunden werden kann.
Diese Ressource behandelt genomweite Analyseverfahren in Forschungsarbeitsabläufen und ist für experimentelles Design, statistische Interpretation und nachgelagerte Priorisierung von Hypothesen gedacht.
Ein starkes genomweites Analyse-Workflow muss daher mehr tun, als Varianten einzeln zu testen. Es muss gleichzeitig drei separate Bedrohungen bewältigen. Die erste ist die Multiplikation. Wenn Millionen von Hypothesen zusammen getestet werden, wird die nominale Signifikanz billig. Die zweite ist die Verwirrung durch Abstammungsstruktur und kryptische Verwandtschaft. Selbst kleine Verzerrungen können in großen Kohorten mächtig werden. Die dritte ist die Kopplungsungleichgewicht. Ein Assoziationspeak kennzeichnet oft einen korrelierten Block, nicht ein einzelnes funktionelles Allel.
Diese drei Bedrohungen definieren auch die drei technischen Schichten, die am wichtigsten sind:
- Fehlerquotensteuerung,
- Strukturkorrektur,
- und kausale Priorisierung.
Wenn eine Schicht schwach ist, wird der Rest des Workflows schwerer zu vertrauen. Deshalb beginnen die nützlichsten modernen GWAS-Diskussionen mit statistischer Strenge, nicht mit biologisch orientierten Erzählungen. Biologie ist wichtig, aber nur nachdem der Assoziationsrahmen Vertrauen gewonnen hat.
Für Projekte, die mit der Generierung neuer Proben anstelle der Wiederverwendung öffentlicher Daten beginnen, beeinflusst die Qualität der upstream-Tests alles, was folgt. Stabile Genotypdaten, konsistente Abdeckung und nachvollziehbare Variantenaufrufe verringern die Unsicherheit in der Folge, bevor das Assoziationsmodell überhaupt beginnt. Je nach Umfang der Studie kann das bedeuten, dass man von Whole-Genome-Sequenzierung, die Analyse von Kohorten im Maßstab mit strukturierten Variantaufrufoder mit Großflächen Whole-Genome-SNP-Genotypisierung wenn die Abdeckung von häufigen Varianten die Hauptpriorität ist.
Statistische Strenge beginnt dort, wo visuelle Signifikanz irreführend wird.
Ein Manhattan-Plot ist überzeugend, weil er Komplexität in Höhe komprimiert. Höhere Spitzen wirken stärker. Dichte Cluster erscheinen überzeugender. Aber das Bild verbirgt eine entscheidende Tatsache: Nicht jedes stark aussehende Signal repräsentiert die gleiche Art von Beweis.
Einige Peaks werden allein durch multiple Tests aufgebläht. Einige spiegeln variationsbedingte Unterschiede wider, die mit der Abstammung korreliert sind, anstatt mit der biologischen Phänotypik. Einige sind echte Assoziationssignale, identifizieren jedoch dennoch nicht die wahre funktionale Variante. Wenn diese Fälle als gleichwertig behandelt werden, wird der Arbeitsablauf visuell klar, aber wissenschaftlich schwach.
Deshalb sollte die statistische Strenge in der genomweiten Analyse als eine Reihe von Filtern und nicht als eine einzelne Schwelle beschrieben werden. Ein Filter steuert, wie viel falsches Signal das Projekt bereit ist zu tolerieren. Ein anderer modelliert, ob die Kohorte selbst das Assoziationsmuster in irreführende Richtungen lenkt. Ein dritter Filter fragt, ob die am höchsten eingestufte Variante tatsächlich der beste Kandidat für die nachgelagerte Validierung ist.
Wenn diese Filter in einer disziplinierten Reihenfolge angewendet werden, wird das Ergebnis interpretierbarer. Das Resultat kann weniger dramatische Behauptungen enthalten, aber die verbleibenden Behauptungen sind viel nützlicher. Dieser Kompromiss ist oft der richtige in Forschungskontexten, insbesondere wenn signifikante Loci später das Fine-Mapping, funktionale Assays oder die Stratifikation von Kohorten leiten.
 Abbildung 1. Statistische Verzerrungen in der genomweiten Analyse entstehen aus verschiedenen Quellen, und jede Korrekturschicht entfernt eine andere Klasse von falschem Vertrauen, bevor die biologische Interpretation beginnt.
Abbildung 1. Statistische Verzerrungen in der genomweiten Analyse entstehen aus verschiedenen Quellen, und jede Korrekturschicht entfernt eine andere Klasse von falschem Vertrauen, bevor die biologische Interpretation beginnt.
Das Mehrfachtestparadoxon in sehr großen Datensätzen
Das Standardproblem der multiplen Tests ist prinzipiell einfach. Wenn eine Studie eine Million Varianten testet, wird selbst eine sehr niedrige nominale Fehlerquote immer noch irreführende Treffer durch Zufall erzeugen. Ein p-Wert, der in einer kleinen Studie überzeugend aussieht, kann in einer genomweiten Untersuchung trivial sein.
Deshalb wurde strikte Schwellenwertsetzung zentral für die Praxis der GWAS. Die Bonferroni-Korrektur ist die klarste Version dieser Logik. Sie teilt das Ziel-alpha durch die Anzahl der Tests und schützt vor familiären Fehlern. Einfach ausgedrückt fragt sie, wie streng die Studie sein muss, wenn bereits ein falsch positives Ergebnis im gesamten Testbereich inakzeptabel ist.
Der Reiz von Bonferroni ist offensichtlich. Es ist transparent. Es ist einfach zu erklären. Es erzeugt eine kurze Liste von Loci, die schwer abzulehnen sind. Wenn die nachgelagerte Validierung teuer ist oder das Projekt um ein sehr konservatives Entdeckungsset herum gestaltet ist, bleibt die Kontrolle im Bonferroni-Stil eine vertretbare Wahl.
Seine Schwäche wird auch offensichtlich, sobald der Suchraum massiv wird. Je strenger die Korrektur, desto mehr echte, aber moderate Signale verschwinden im Rauschen. Dies schafft das zentrale Paradoxon der multiplen Tests in der genomweiten Analyse: Eine breitere Suche erhöht die Chance, echte biologische Effekte zu entdecken, aber die erforderliche Schwelle zur Kontrolle von falsch positiven Ergebnissen kann so streng werden, dass sie gleichzeitig schwächere echte Effekte unterdrückt.
Ansätze zur Kontrolle der falschen Entdeckungsrate nähern sich demselben Problem aus einem anderen Blickwinkel. Anstatt zu fragen, wie man überhaupt falsche Positive vermeiden kann, fragt die FDR, welcher Anteil der als Entdeckungen bezeichneten Ergebnisse als falsch toleriert werden kann. Diese Verschiebung verändert den Zweck des Schwellenwerts.
Bonferroni eignet sich am besten für bestätigungsorientierte EntdeckungFDR ist oft besser geeignet für kandidatenbewahrende Entdeckung.
Das macht FDR nicht nachlässig. Es macht es zielbewusst. In vielen realen GWAS-Workflows besteht das Ziel nicht darin, eine endgültige, unveränderliche Liste von Loci zu erstellen. Das Ziel ist es, einen bedeutungsvollen Kandidatenraum zu bewahren, der dann durch Replikation, Feinabstimmung, Kointegration und funktionale Integration eingegrenzt werden kann. In diesem Kontext kann FDR der praktischere erste Ansatz sein.
Der Fehler besteht darin, diese Methoden als moralische Gegensätze zu betrachten. Das sind sie nicht. Sie beantworten unterschiedliche Fragen:
- Bonferroni fragt, wie man sich gegen falsche Positive in der getesteten Familie schützen kann.
- FDR fragt, wie der erwartete Anteil falscher Entdeckungen unter den beibehaltenen Treffern verwaltet werden kann.
- Bonferroni bevorzugt kurze, schwer zu widerlegende Listen.
- FDR befürwortet umfassendere Entdeckungsstufen, die für nachgelagertes Kürzen offen bleiben.
In fortgeschrittenen Projekten ist die beste Lösung oft, beide Ideen in verschiedenen Phasen zu verwenden. Ein Schwellenwert definiert die strikte Kernassoziationsschicht. Ein anderer bewahrt einen breiteren Kandidatensatz für Feinabstimmung und mechanismusorientierte Nachverfolgung. Dies ist besonders nützlich, wenn das Studiendesign nicht nur auf der Spitzenberichterstattung basiert, sondern auf kausaler Priorisierung.
Die praktische Lektion ist einfach: Signifikanz ist kein einziger universeller Zustand. Sie hängt davon ab, wie das Projekt Fehler definiert, was es mit den beibehaltenen Loci plant und wie viel Unsicherheit es bereit ist, in die nächste Phase mitzunehmen.
Die Populationsstratifizierung ist kein geringfügiges Kovariatenproblem.
Die Populationsstratifizierung wird oft als Störfaktor eingeführt. Diese Formulierung ist zu milde. In großen genomweiten Studien stellt sie eine strukturelle Bedrohung dar.
Das Problem tritt auf, wenn die Allelfrequenzen zwischen Subgruppen variieren und diese Subgruppen auch in der Phänotyp-Prävalenz aus Gründen unterscheiden, die nicht mit der untersuchten ursächlichen Variante zusammenhängen. Wenn diese Struktur nicht richtig behandelt wird, kann das Modell die Kohortenkomposition mit der Biologie verwechseln. Das resultierende Signal kann stabil, statistisch stark und biologisch plausibel erscheinen, während es dennoch von Störfaktoren beeinflusst wird.
Dies ist ein Grund, warum einige Assoziationsgipfel zusammenbrechen, wenn sich das Kohortendesign ändert, wenn sich die Abstammungszusammensetzung verschiebt oder wenn rigorosere Strukturkorrekturen angewendet werden. Das Problem ist nicht, dass die Analyse an Power mangelte. Das Problem ist, dass das Modell der strukturierten Variation zu viel Bedeutung beimaß.
Die Hauptkomponentenanalyse bleibt eines der nützlichsten Werkzeuge zur Diagnose und Anpassung der Abstammungsstruktur. PCA komprimiert die Hauptachsen der Variation in kontinuierliche Komponenten, die als feste Kovariaten hinzugefügt werden können. Sie ist rechnerisch effizient, interpretierbar und nach wie vor äußerst wertvoll für die explorative Kohortenbewertung. In vielen Datensätzen bewältigt sie die breite Struktur ausreichend gut, um die Kalibrierung erheblich zu verbessern.
Aber PCA hat klare Grenzen.
PCA erfasst dominante Variationsachsen. Es modelliert nicht vollständig die gesamte Stichprobenkovarianz. Es absorbiert nicht vollständig kryptische Verwandtschaft. Es stellt nicht vollständig eine verteilte verwandtschaftsähnliche Struktur dar, die bestehen bleiben kann, nachdem breite Abstammungstrends entfernt wurden. In moderaten und großen Kohorten, insbesondere in solchen mit subtiler Familienstruktur oder heterogenem Sampling-Verlauf, kann residuale Verfälschung die PCA-einzige Korrektur überstehen.
Dort werden lineare Mischmodelle wichtig.
Warum lineare gemischte Modelle die moderne GWAS-Praxis verändert haben
Ein lineares gemischtes Modell fügt eine Zufallseffektkomponente hinzu, die die Kovarianz zwischen Individuen erfasst, oft durch eine genetische Verwandtschaftsmatrix oder eine eng verwandte Darstellung. Dies verändert die Logik der Korrektur.
PCA sagt: Hauptstrukturachsen herausrechnen.
LMM sagt: Modellierte den korrelierten Hintergrund direkt.
Dieser Unterschied ist nicht kosmetisch. Er ist der Grund, warum die gemischte Modellassoziation in großen, strukturierten Kohorten zentral wurde. Anstatt sich nur auf eine Handvoll fester Kovariaten zu verlassen, erkennt das Modell, dass Individuen genetische Ähnlichkeiten im Hintergrund teilen können, die die Assoziationsstatistiken über das Genom hinweg beeinflussen.
Dies macht LMM besonders wertvoll, wenn:
- kryptische Verwandtschaft ist wahrscheinlich,
- subtile Verwandtschaft bleibt nach grundlegender Qualitätskontrolle bestehen,
- Die Kohortengröße ist groß genug, dass schwache Störfaktoren hochsignifikant werden.
- Die Probenstruktur ist diffus und nicht klar getrennt.
- oder die nachgelagerte Interpretation hängt von marginalen Signalen ab, die anfällig für Inflation wären.
In diesen Einstellungen ist die gemischte Modellassoziation kein Luxusmerkmal. Sie ist Teil des grundlegenden inferenziellen Designs.
Das bedeutet nicht, dass PCA irrelevant wird. Gute Arbeitsabläufe nutzen oft beides. PCA bleibt nützlich für die Visualisierung von Abstammung, die Erkennung von Ausreißern, die explorative Bewertung von Proben und die Modellierung von festen Effekten. LMM fügt dann eine stärkere zweite Schutzschicht während des Assoziationstests selbst hinzu. Das eine hilft, die Kohorte zu beschreiben. Das andere hilft, die aus ihr gezogenen Schlussfolgerungen zu stabilisieren.
Hier wird auch die Softwarewahl bedeutend. Ein standardisierter, regressionsbasierter Workflow kann in einer Kohorte vollständig ausreichend und in einer anderen unzureichend sein. Die Entscheidung sollte der Probenarchitektur folgen, nicht den Gewohnheiten des Analytikers. Für locus-fokussierte Nachverfolgung nach breiter Entdeckung bewegen sich einige Projekte auch in engere Assay-Designs wie gezielte Regionssequenzierung oder eine benutzerdefinierte SNP-Fine-Mapping Workflow, nachdem der breitere Assoziationsbereich bereits reduziert wurde.
Wie man erkennt, wann eine PCA-Only-Korrektur nicht ausreicht.
Viele Studien beinhalten Hauptkomponenten, da dieser Schritt Standard ist. Weniger Studien erklären, warum die gewählte Korrekturstrategie für diese Kohorte ausreichend war. Dort kann eine stärkere technische Schreibweise Mehrwert schaffen.
Die Korrektur nur mit PCA kann ausreichend sein, wenn die Kohorte relativ sauber ist, die Verwandtschaft begrenzt ist, die Struktur breit und nicht tief verschachtelt ist und das Projekt nicht stark auf Grenzsignale angewiesen ist. Sie wird weniger beruhigend, wenn der Datensatz groß ist, die Rekrutierung heterogen ist oder versteckte Kovarianzmuster plausibel sind.
Die Frage ist nicht, ob PCs einbezogen wurden. Die Frage ist, ob das Strukturproblem tatsächlich gelöst wurde.
Mehrere Warnsignale sollten Vorsicht hervorrufen:
- residuelle Inflation nach standardmäßiger Korrektur,
- Assoziationsverschiebungen, die die Abstammungszusammensetzung verfolgen,
- unerwartete Persistenz eines schwachen genomweiten Signals,
- starke Auswirkungen in Regionen, die für ihre Empfindlichkeit gegenüber Strukturen bekannt sind,
- oder instabile Ergebnisse über verwandte, aber unterschiedlich gefilterte Kohortenuntergruppen.
Diese Signale beweisen nicht automatisch, dass PCA gescheitert ist. Sie deuten darauf hin, dass das Projekt möglicherweise ein stärkeres Kovarianzmodell benötigt.
Die umfassendere Lehre ist es wert, klar ausgesprochen zu werden: Die Bevölkerungsanpassung sollte entworfen und nicht übernommen werden. Zu viele GWAS-Pipelines verwenden immer noch das Struktur-Anpassungsrezept des letzten Projekts mit minimaler Rechtfertigung. Das ist riskant bei Daten im Maßstab von 2026, wo subtile Störfaktoren statistisch verstärkt werden können, lange bevor sie visuell offensichtlich werden.
Der LD-Flaschenhals beginnt dort, wo viele GWAS-Zusammenfassungen enden.
Sobald die Assoziationstests abgeschlossen sind, springen viele Leser zum signifikantesten SNP und fragen, welche Variante den Phänotypwechsel verursacht hat. Diese Frage ist verständlich. Sie ist jedoch in der Regel auch verfrüht.
Der führende SNP ist die Variante mit der stärksten Assoziationsstatistik in den getesteten Daten. Es ist nicht automatisch die Variante, die die Expression ändert, das Spleißen beeinflusst, Chromatin stört oder direkt die phänotypische Biologie antreibt. In vielen Loci ist der führende SNP einfach das beste statistische Tag für ein nahegelegenes kausales Allel, da mehrere Varianten durch Linkage-Disequilibrium korreliert sind.
Das ist der LD-Flaschenhals.
Die Assoziation erkennt eine Region. Die Biologie benötigt eine Variante. Die Lücke zwischen diesen beiden Ebenen ist genau der Punkt, an dem viele oberflächliche GWAS-Interpretationen über das Ziel hinausschießen.
In einem Locus mit starker LD können mehrere benachbarte Varianten gemeinsam ansteigen. Ihre p-Werte können ähnlich sein. Ihre Rangordnung kann sich über Abstammungsgruppen, Imputationspanels oder Kohortendesigns hinweg verschieben. Diese Instabilität ist kein technisches Ärgernis. Sie ist ein Hinweis. Sie sagt dem Analysten, dass das Signal ein korreliertes Umfeld darstellt und nicht einen einzelnen gelösten Mechanismus.
Ein ausgereifter Workflow betrachtet daher den führenden SNP als Einstiegspunkt, nicht als endgültige Antwort. Dies ist besonders wichtig, wenn erwartet wird, dass die Studie nachgelagerte Störungsarbeiten, Ausdrucksnachverfolgung oder regulatorische Validierung unterstützt. Experimentelle Teams benötigen nicht den lautesten SNP. Sie benötigen das am besten verteidigbare Kandidatenset.
Dieser Bedarf drängt das Projekt oft über reine Assoziationsdaten hinaus und hin zu funktionsorientierten Assays. Wenn das Ziel darin besteht, locus-spezifische Statistiken mit regulatorischen Mechanismen zu verbinden, können Teams Assoziationsbefunde mit RNA-Seqgezielte Chromatin-Profilierung wie ATAC-Seqoder breiter koordiniert Multi-Omics-Dienstleistung Unterstützung bei der Bestimmung, ob priorisierte Varianten in einem plausiblen regulatorischen Kontext liegen.
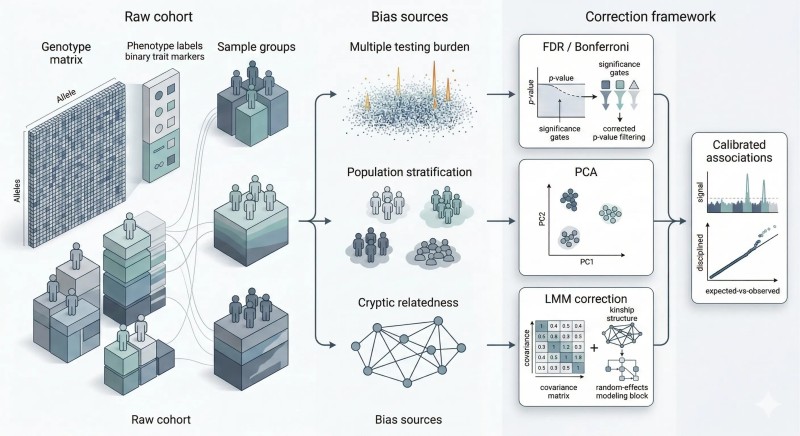 Abbildung 2. Ein Assoziationspeak repräsentiert normalerweise ein durch LD definiertes Umfeld und nicht eine einzelne mechanistische Antwort, weshalb die Rangfolge der Haupt-SNPs von einer Priorisierung der glaubwürdigen Sets gefolgt werden muss.
Abbildung 2. Ein Assoziationspeak repräsentiert normalerweise ein durch LD definiertes Umfeld und nicht eine einzelne mechanistische Antwort, weshalb die Rangfolge der Haupt-SNPs von einer Priorisierung der glaubwürdigen Sets gefolgt werden muss.
Feinabgleich ist die eigentliche Brücke zwischen Assoziation und Kausalität.
Feinabgleich existiert, weil GWAS und Mechanismen auf unterschiedlichen Auflösungen arbeiten. GWAS ist darauf optimiert, Loci zu identifizieren, die mit einem Phänotyp assoziiert sind. Feinabgleich ist darauf optimiert zu entscheiden, welche Varianten innerhalb dieses Locus nach Berücksichtigung der LD-Struktur weiterhin Glauben verdienen.
Diese Unterscheidung ist grundlegend.
Eine nützliche Möglichkeit, die Beziehung zu betrachten, ist folgende:
- GWAS fragt, welcher Locus wichtig ist.
- Feinabgleich fragt, welche Varianten innerhalb dieses Lokus plausible ursächliche Kandidaten bleiben.
Sobald es so formuliert wird, wird der Bedarf an Feinabstimmung offensichtlich. Allein das Assoziationsranking kann eine kausale Frage nicht beantworten, wenn viele korrelierte Varianten gemeinsam auftreten.
Der frequentistische Ansatz zur Verfeinerung nähert sich diesem Problem oft durch bedingte Tests und iterative Signifikanzbewertung. Das kann helfen festzustellen, ob der Ort mehrere unabhängige Signale enthält. Es bleibt nützlich. Aber es neigt immer noch dazu, die Sprache der Schwellenüberlebensanalyse zu sprechen.
Bayesian Fine-Mapping verändert die Diskussion, indem sie fragt, wie die Unterstützung auf die Kandidatenvarianten und die potenziellen ursächlichen Konfigurationen verteilt werden sollte. Anstatt nur zu fragen, ob eine Variante nach der Bedingung signifikant bleibt, wird gefragt, wie viel posteriorer Glaube jeder Kandidat basierend auf dem beobachteten Muster und der lokalen LD-Struktur erhalten sollte.
Dieser Wandel ist kraftvoll, da Experimente teuer sind. Die meisten Teams können nicht jede Variante in einem zugehörigen Block testen. Sie benötigen eine rangierte, unsicherheitsbewusste Shortlist. Bayesian Fine-Mapping bietet genau das.
Eine posteriori Einschlusswahrscheinlichkeit ist keine Garantie für die Wahrheit. Ein glaubwürdiges Set ist kein Versprechen, dass die kausale Variante mit Sicherheit erfasst wurde. Aber beides ist weitaus ehrlicher und operationell nützlicher, als vorzugeben, dass das stärkste Assoziationssignal bereits den Mechanismus gelöst hat.
Dies verbessert auch den Übergang zwischen den rechnerischen und experimentellen Teams. Ein schwacher Workflow sendet einen SNP mit zu viel Vertrauen nach unten. Ein stärkerer Workflow sendet ein rangiertes Kandidatenset, erklärt, warum Unsicherheit bleibt, und klärt, welche Art von funktionalen Beweisen diese Unsicherheit weiter verringern würde.
Hier beginnt der zweite Teil des Artikels. Sobald ein Locus präzise in einen glaubwürdigen Kandidatenbereich eingegrenzt wurde, ist die nächste Frage nicht mehr, welche Region assoziiert ist. Die nächste Frage ist, wie diese Kandidatenvarianten mit der Expression, dem Spleißen, dem regulatorischen Zustand und schließlich mit kohortenbasierten Architekturen wie polygenetischen Risikoscores verbunden sind.
Multi-omische Integration verwandelt assoziierte Loci in biologische Hypothesen.
Das Fine-Mapping verengt den Kandidatenraum. Es vervollständigt jedoch nicht die biologische Geschichte.
Ein glaubwürdiges Set ist immer noch ein statistisches Objekt. Es zeigt uns, welche Varianten nach LD-bewusster Modellierung plausibel bleiben. Es sagt uns jedoch noch nicht, wie diese Varianten wirken, welcher Gewebekontext am wichtigsten ist, ob der Haupteffekt auf die Expression oder das Spleißen wirkt oder welches Gen in der Region das wahre Effektor-Gen ist. An diesem Punkt wird die Integration von Multi-Omics notwendig.
Die schwächste Version dieses Schrittes ist einfache Überlappung. Eine Studie identifiziert ein GWAS-Lokus, stellt fest, dass sich in derselben Region ein eQTL befindet, und weist dann das nahegelegene Gen als den wahrscheinlichsten Mechanismus zu. Dieser Ansatz ist verbreitet, da er schnell und einfach zu erklären ist. Er ist jedoch oft unvollständig. Viele Loci lassen sich nicht eindeutig nur durch Expressionsdaten aufklären, und einige werden besser durch Splicing, Chromatinzugänglichkeit oder regulatorischen Kontext erklärt, der in allgemeinen eQTL-Zusammenfassungen nicht sichtbar ist. Neuere Arbeiten unterstützen weiterhin die Idee, dass eine mehrschichtige QTL-Interpretation Mechanismen aufdecken kann, die bei einer ausschließlich eQTL-basierten Betrachtung übersehen würden.
Deshalb sollte die ernsthafte Interpretation nach GWAS als folgendes formuliert werden: kausale Triangulation, keine Anmerkung.
Ein robuster Triangulationsworkflow stellt eine verknüpfte Reihe von Fragen:
- Kollidiert das glaubwürdige Set mit einem eQTL-Signal?
- Ändert dieselbe Region die Transkriptstruktur durch einen sQTL-Effekt?
- Befindet sich die Kandidatenvariante in offener Chromatinstruktur oder einem anderen aktiven regulatorischen Element?
- Hat das betroffene Gen biologischen Sinn im Gewebe oder Zelltyp, der für den Phänotyp relevant ist?
- Deuten mehrere unabhängige Datenebenen auf denselben Mechanismus hin, oder stehen sie im Widerspruch zueinander?
Je stärker die Konvergenz, desto stärker die Hypothese.
eQTL ist nützlich, aber es ist nicht die vollständige Antwort.
Expression-QTLs bleiben eine der wertvollsten Brücken zwischen Genotyp und Funktion. Sie können erklären, warum ein nicht-kodierendes Signal wichtig ist, helfen, Effektor-Gene zu priorisieren, und lenken die Diskussion von Annahmen über das nächstgelegene Gen weg. Aber sie haben Grenzen, die klar benannt werden müssen.
Zuerst sind eQTL-Effekte kontextabhängig. Eine Variante kann die Expression in einem Gewebe regulieren und in einem anderen nicht. Sie kann nur in einem Entwicklungsfenster, unter einem Stimulationszustand oder in einem seltenen Zelltyp wirken, den die Daten des Gesamtgewebes nicht auflösen können. Zweitens ist die Gesamtexpression nur ein Ergebnis. Einige Varianten verändern das Isoform-Gleichgewicht, die Exon-Einschlussrate oder die Transkriptverwendung, ohne eine große Verschiebung der Gesamtexpression zu erzeugen. Drittens beweist ein gemeinsames regionales Signal nicht eine gemeinsame Kausalität. Ein GWAS-Peak und ein eQTL-Peak können im selben LD-Block überlappen, während sie dennoch von unterschiedlichen zugrunde liegenden Varianten angetrieben werden.
Hier wird der sQTL-Beweis besonders wertvoll. Ein Locus, der im eQTL-Raum bescheiden aussieht, kann viel überzeugender werden, wenn splicing-sensible Daten berücksichtigt werden. Aus diesem Grund wird die Interpretation nach GWAS oft viel stärker, wenn die standardmäßige Transkriptom-Profilierung mit isoformauflösenden oder transkriptstrukturbewussten Workflows kombiniert wird.
In praktischen Forschungsumgebungen kann das bedeuten, dass man kombiniert RNA-Seq mit Vollständige Transkript-Sequenzierung (Iso-Seq) wenn die Isoform-Architektur wichtig ist, oder hinzufügen ATAC-Seq wenn die regulatorische Zugänglichkeit Teil der Hypothese ist. Wenn der Mechanismus wahrscheinlich über mehrere molekulare Schichten verteilt ist, ein koordiniertes Multi-Omics-Dienstleistung Der Rahmen kann informativer sein als eine Einzeluntersuchung. Diese Dienstanweisungen stammen aus der Datei, die Sie bereitgestellt haben.
Kollokalisierung ist strenger als Überlappung.
Einer der häufigsten Fehler bei der Interpretation von GWAS ist es, genomische Nähe als mechanistische Evidenz zu behandeln. Der Locus überlappt ein eQTL, daher ist das Gen ursächlich. Dieser Schritt ist zu schnell.
Die Kolokalisation stellt eine viel strengere Frage: Sind das GWAS-Signal und das molekulare QTL-Signal mit demselben zugrunde liegenden kausalen Variant konsistent, oder sind sie einfach benachbarte Signale innerhalb desselben LD-Blocks? Diese Unterscheidung ist wichtig, da Überlappungen ohne Kolokalisation falsche narrative Gewissheit schaffen können.
Eine starke Interpretationskette sieht daher folgendermaßen aus:
- den zugehörigen Locus erkennen,
- feinabstimmen des glaubwürdigen Kandidatensatzes,
- Testen Sie die Koinlokalisierung mit eQTL- oder sQTL-Daten.
- bewerten Sie die Relevanz von Gewebe und Zelltyp,
- integriere Chromatin- oder regulatorische Beweise,
- priorisiere das am besten verteidigbare Effektor-Gen oder regulatorische Mechanismus.
Das ist langsamer als die Zuordnung des nächstgelegenen Gens. Es ist auch viel glaubwürdiger.
Die Multi-Omik-Integration sollte die Unsicherheit verringern, nicht das Ergebnis schmücken.
Es gibt hier ein subtil, aber wichtiges Prinzip. Mehr Daten bedeuten nicht automatisch mehr Schlussfolgerungen. Die Integration von Multi-Omics ist nur dann wertvoll, wenn sie die Unsicherheit verringert.
Wenn eQTL, sQTL, offene Chromatin und der Kontext von Signalwegen alle auf dasselbe Gen oder regulatorische Ereignis konvergieren, steigt das Vertrauen. Wenn diese Ebenen nicht übereinstimmen, ist das Ergebnis kein Misserfolg. Es ist eine nützliche Einschränkung. Das Projekt hat gelernt, dass der Mechanismus noch ungelöst ist und dass eine gezielte Validierung entsprechend entworfen werden sollte.
Das ist die richtige Denkweise für eine fortgeschrittene genomweite Analyse. Das Ziel ist nicht, die überladenste Abbildung zu erstellen. Das Ziel ist es, mit den wenigsten ungerechtfertigten Sprüngen von der Assoziation zum Mechanismus zu gelangen.
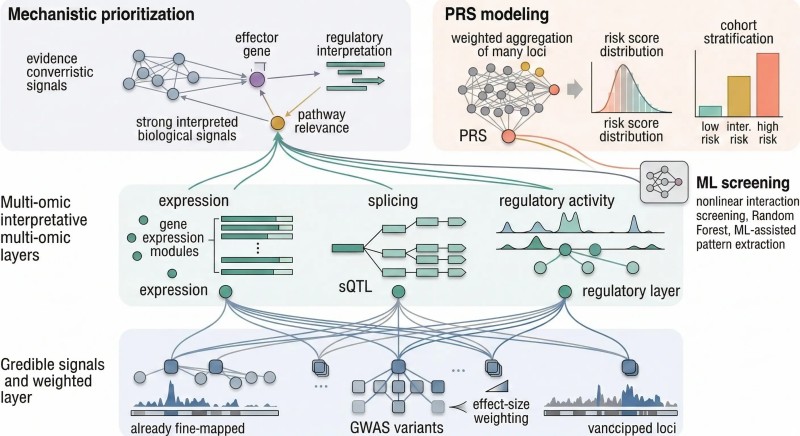 Abbildung 3. Die multi-omische Interpretation ist am stärksten, wenn mehrere funktionale Ebenen auf denselben Kandidatenmechanismus konvergieren, während PRS diese statistisch fundierten Loci verwendet, um das Signal auf Kohortenebene zu modellieren, anstatt die Kausalität auf Einzel-Locus-Ebene.
Abbildung 3. Die multi-omische Interpretation ist am stärksten, wenn mehrere funktionale Ebenen auf denselben Kandidatenmechanismus konvergieren, während PRS diese statistisch fundierten Loci verwendet, um das Signal auf Kohortenebene zu modellieren, anstatt die Kausalität auf Einzel-Locus-Ebene.
Polygenetische Risikoscores sind ein Aggregationsproblem, das auf strenger Vorarbeit basiert.
Sobald die Analyse über einzelne Loci hinausgeht, besteht die nächste Versuchung darin, die Architektur in einen einzigen Wert zu komprimieren. Polygenetische Risikoscores tun genau das. Sie aggregieren gewichtete Effekte über viele Loci, um das verteilte vererbte Signal auf Kohortenebene zu modellieren.
Das ist nützlich. Es ist auch leicht, es missbräuchlich zu verwenden.
Ein PRS erbt die Stärken und Schwächen jeder vorhergehenden Stufe. Wenn die Assoziationsschicht voreingenommen ist, erbt der Score diese Voreingenommenheit. Wenn die Abstammungsstruktur schlecht behandelt wird, leidet die Übertragbarkeit. Wenn LD nachlässig modelliert wird, kann der Score instabil sein. Wenn Effektgrößen in einer Population geschätzt werden, die nicht mit der Zielkohorte übereinstimmt, kann die Leistung stark abnehmen. Neuere Übersichten und Methodenschriften betonen weiterhin, dass die Genauigkeit von PRS stark von Abstammung, LD-Handhabung, Modellprioren und der Art und Weise, wie die Schrumpfung der Effektgrößen implementiert wird, geprägt ist.
Was PRS gut in Forschungsgruppen macht
PRS ist am nützlichsten, wenn es als Modell für verteilte Signale betrachtet wird, anstatt als Abkürzung für mechanistische Erklärungen.
In Forschungsabläufen kann PRS helfen:
- Stichproben in belastungsdefinierte Gruppen schichten,
- testen, ob das Signal diffus oder konzentriert ist,
- Cohorten für nachgelagerte Vergleiche anreichern,
- Vergleiche Architektur über verwandte Merkmale.
- und bieten einen kohortenbasierten Ergänzung zu locus-basierten biologischen Ansätzen.
Diese Einordnung ist wichtig. PRS beantwortet eine andere Frage als das Fine-Mapping. Fine-Mapping fragt, welche Varianten innerhalb eines Lokus plausible kausale Kandidaten bleiben. PRS fragt, wie viele gewichtete Loci zusammen genommen die Varianz in der Kohorte erklären.
Diese sind keine konkurrierenden Ziele. Sie operieren auf unterschiedlichen Auflösungsebenen.
Die eigentliche Herausforderung besteht nicht in der Summation. Es ist das Gewichtung.
Auf den ersten Blick scheint PRS einfach zu sein. Zähle die Allele. Gewichte sie nach Effektgröße. Summiere über die Loci. Aber fast jeder Teil dieses Satzes verbirgt eine Modellierungsentscheidung.
- Welche Loci sind enthalten?
- Werden nur genomweit signifikante Loci verwendet?
- Werden Subthreshold-Varianten beibehalten?
- Wie wird LD behandelt?
- Werden Effektgrößen geschrumpft?
- Werden funktionale Annotationen verwendet, um Gewichtungen zu informieren?
- Ist der Score in einer an die Abstammung angepassten Population kalibriert?
Jede dieser Entscheidungen beeinflusst das Endergebnis.
Ein Score, der nur aus Top-Signalen besteht, ist leichter zu erklären, kann jedoch diffuse Architekturen übersehen. Ein breiterer Score kann mehr Varianz erfassen, kann jedoch auch mehr Rauschen importieren, wenn LD-Pruning, Schrumpfung oder Ahnenabgleich schwach sind. Annotation-informierte Modelle versuchen, einen Teil dieses Problems zu lösen, indem sie biologische Prämissen verwenden, um Varianten, die wahrscheinlich funktional bedeutsam sind, stärker zu gewichten. Diese Richtung wird zunehmend attraktiver, da Forscher versuchen, prädiktive Modellierung mit mechanistischer Plausibilität zu kombinieren.
PRS sollte ein gutes Verbandsdesign unterstützen, nicht ersetzen.
Eine der einfachsten Möglichkeiten, einen GWAS-Artikel zu schwächen, besteht darin, PRS als einen Aufstiegspfad erscheinen zu lassen, der die rigorose Analyse auf Locus-Ebene umgeht. Das ist es nicht.
PRS ist am stärksten, wenn es auf einem guten Assoziationsdesign, einer guten Strukturkorrektur und einer guten Lokusinterpretation basiert. In einem ausgereiften Workflow:
- Vereinigung legt fest, welche Regionen wichtig sind,
- Feinabgleich schränkt die Kandidatenvarianten ein,
- multi-omische Daten klären plausible Funktionen,
- PRS aggregiert verteilte Effekte über die Kohorte.
Das ist die richtige Reihenfolge der Ideen.
Für Teams, die die Konstruktion von Kohorten-Scores in großem Maßstab planen, ist die Wahl der Plattform ebenfalls wichtig. Je nach Architektur, Budget und gewünschter Dichte des Standorts kann die Datenquelle upstream stammen von Whole-Exom-Sequenzierung, Humane/Maus Whole-Exom-Sequenzierung, SNP-Mikroarrayoder Genotypisierung durch Sequenzierung (GBS)Diese Optionen stammen aus dem von Ihnen bereitgestellten Serviceinventar und passen zu verschiedenen Forschungsmaßstab-PRS-Designs.
Maschinelles Lernen für Epistase ist wertvoll, aber hauptsächlich als Screening-Ebene.
Maschinelles Lernen tritt in die genomweite Analyse aus einem einfachen Grund ein. Klassische GWAS sind am stärksten für additive Effekte, die Marker für Marker getestet werden. Biologie ist jedoch nicht immer additiv. Gen-Gen-Interaktionen, Schwellenverhalten und nichtlineare Kombinationen können von Bedeutung sein. Random Forests und verwandte Methoden sind daher attraktiv, da sie nach Interaktionsmustern suchen können, die gewöhnliche marginale Assoziationen übersehen können.
Dieses Versprechen ist real. Die häufige Übertreibung ist, dass maschinelles Lernen daher klassische GWAS ersetzt. Das tut es nicht.
Jüngste Arbeiten zur polygenen Vorhersage zeigen weiterhin, dass komplexere Modelle nicht automatisch bessere Ergebnisse als starke lineare oder gemischte Modell-Baselines liefern. In vielen Fällen ist der erwartete Gewinn durch Nichtlinearität geringer als behauptet, und einige berichtete Verbesserungen verringern sich, wenn die Benchmarking-Methoden strenger werden.
Das macht maschinelles Lernen nicht irrelevant. Es definiert seine richtige Rolle.
Wo Random Forests und verwandte Modelle echten Mehrwert bieten
Maschinenlernmodelle sind nützlich, wenn die Forschungsfrage explorativ ist:
- Gibt es potenzielle nichtlineare Wechselwirkungen, die es wert sind, getestet zu werden?
- Teilen bestimmte Variantenkombinationen die Kohorte auf unerwartete Weise?
- Gibt es hochrangige Merkmalsmuster, die eine gezielte Nachverfolgung verdienen?
In diesem Kontext fungiert maschinelles Lernen als ein Screening-ToolEs schlägt Kandidaten für eine tiefere Analyse vor. Es ersetzt nicht den statistischen Rahmen, der ursprünglich die zugrunde liegende Glaubwürdigkeit festgelegt hat.
Diese Rolle ist besonders sinnvoll für Epistase-Arbeiten. Der gesamte Interaktionsraum ist enorm. Eine gut gestaltete ML-Phase kann helfen, die Suche auf Muster einzugrenzen, die eine formale Bewertung wert sind, aber nur, wenn der Workflow bereits eine disziplinierte Vorverarbeitung, Abstammungskontrolle und ein starkes Basismodell zum Vergleich aufweist.
Die drei Hauptfallen bei der ML-basierten Epistase-Analyse
Die erste Falle ist Feature-ExplosionDie Anzahl möglicher Interaktionen wächst schnell, und die meisten davon sind uninformativ. Ohne vorherige Filterung investiert das Modell zu viel Aufwand in Rauschen.
Die zweite Falle ist InterpretierbarkeitsverlustEine prädiktive Struktur kann real sein, ohne mechanistisch informativ zu sein. Ein Modell kann auch ahnenkorrelierte oder LD-redundante Muster lernen, die biologisch interessant erscheinen, es aber nicht sind.
Die dritte Falle ist schwache BenchmarkingEin komplexes Modell wirkt nur beeindruckend, wenn die Basis unzureichend ist. Der richtige Vergleich erfolgt nicht mit einem simplen, zufällig erstellten additiven Modell. Er erfolgt mit einer starken, LD-bewussten, ahnenbewussten Basis, die gut aufgebaut ist.
Deshalb sollte maschinelles Lernen normalerweise spät im Workflow eingesetzt werden. Es fügt den größten Wert hinzu, nachdem die Studie bereits eine stabile Assoziationsstruktur und glaubwürdige Kandidatenloci etabliert hat.
Die Softwarewahl sollte der Kohortenstruktur folgen, nicht den Gewohnheiten.
Viele Zusammenfassungen erwähnen PLINK, BOLT-LMM und REGENIE im gleichen Atemzug, als wären sie austauschbar. Das sind sie nicht. Sie überschneiden sich in ihrem Zweck, lösen jedoch unterschiedliche Probleme mit unterschiedlichen Stärken. Die offizielle Dokumentation macht das deutlich: PLINK 2.0 betont schnelle Standard-Assoziations-Workflows, BOLT-LMM konzentriert sich auf gemischte Modellassoziationen in großen Kohorten, und REGENIE ist für skalierbare Whole-Genome-Regressionsanalysen im modernen Kohortenskalierungsmaßstab konzipiert.
Vergleich von GWAS-Software
| Software | Hauptstärke | Geschwindigkeitsprofil | Speicherprofil | Verwandtschafts- / Strukturverwaltung | Bestmöglicher Anwendungsfall | Hauptvorsicht |
|---|---|---|---|---|---|---|
| PLINK 2.0 | Schnelle Baseline-Zuordnung, QC-intensive Arbeitsabläufe, transparente Regressionskonfiguration | Schnell für Standard-Regression-Workflows | Moderat | Verlässt sich normalerweise auf PCA/Kovariatenkorrektur anstelle der vollständigen Handhabung von gemischten Modellstrukturen. | Saubere oder moderat strukturierte Kohorten, schnelle Screening, standardisierte additive Analyse | Kann allein möglicherweise unzureichend sein, wenn subtile Zusammenhänge oder großflächige Strukturen im Mittelpunkt stehen. |
| BOLT-LMM | Gemischtes Modell der Assoziation in großen Kohorten mit verteilter Verwandtschaft | Hoch, einmal konfiguriert für große menschliche Datensätze | Mäßig bis hoch | Starke LMM-basierte Handhabung von Verwandtschaft und Hintergrundstruktur | Große menschliche Kohorten mit subtiler Struktur und polygenem Hintergrund | Erfordert eine sorgfältige Bewertung der Eignung der Kohorte und Aufmerksamkeit für das Gleichgewicht zwischen Fall und Kontrolle. |
| REGENIE | Skalierbare Ganzgenomregression für sehr große Datensätze und viele Merkmale | Sehr hoch in großen modernen Rohrleitungen | Effizient im Verhältnis zur Skalierung | Stark für große strukturierte Datensätze und Hochdurchsatz-Assoziationstests | Biobank-große Workflows, viele Phänotypen, große binäre oder quantitative Merkmalsstudien | Der zweistufige Workflow erhöht die Komplexität der Einrichtung und hängt von einer disziplinierten Eingabevorbereitung ab. |
Dies ist kein Gewinner-alles-oder-nichts-Tisch. Es ist ein Zuordnungstisch.
Wie man in der Praxis wählt
Wählen PLINK wenn die Hauptpriorität Geschwindigkeit, transparente Basislinienassoziation, starke QC-Integration und eine Kohorte sind, bei der subtile Verwandtschaft nicht die Hauptbedrohung für die Inferenz darstellt.
Wählen BOLT-LMM wenn das Projekt von einer gemischten Modellkorrektur in einer großen menschlichen Kohorte mit verteilter Verwandtschaft und polygenem Hintergrund abhängt.
Wählen REGENIE wenn Skalierung, Durchsatz und effiziente Assoziationen in großen Kohorten am wichtigsten sind, insbesondere wenn das Projekt viele Merkmale oder umfangreiche Analysen binärer Merkmale durchführen muss.
Die beste Softwarewahl ist immer an die Kohortenarchitektur gebunden. Es geht niemals nur um Beliebtheit.
Wie eine fortgeschrittene genomweite Analyse jetzt aussehen sollte
Ein ausgereiftes genomweites Analyse-Workflow sollte nicht bei der Signifikanz haltmachen, und es sollte Assoziation, Mechanismus und Vorhersage nicht in eine einzige Behauptung zusammenfassen.
Ein stärkeres Betriebsmodell sieht so aus:
- stabile Variantendaten generieren oder kuratieren,
- Wählen Sie eine Fehlerkontrollstrategie, die zu den Projektzielen passt.
- Modellieren Sie Abstammung und Verwandtschaft rigoros,
- Behandle führende SNPs als Ausgangspunkte und nicht als Schlussfolgerungen.
- fein-mappen Loci unter LD-bewusster Unsicherheit,
- mechanistische Hypothesen durch eQTL, sQTL und regulatorische Integration testen,
- Verwenden Sie PRS, um die verteilte Architektur auf Kohortenebene zusammenzufassen.
- maschinelles Lernen selektiv für die Interaktionsscreening anwenden,
- Wählen Sie Software entsprechend der Skalierung und Struktur aus.
- Kommunizieren Sie Unsicherheiten klar bei jedem Übergang.
Diese Reihenfolge ist wichtig, da jede Phase eine andere Frage beantwortet. Die Assoziation fragt, wo das Signal ist. Das Fine-Mapping fragt, welche Varianten plausibel bleiben. Die multi-omische Integration fragt, wie das Signal wirken könnte. PRS fragt, wie das Signal in der Kohorte akkumuliert. Maschinelles Lernen fragt, ob höherordentliche Muster eine genauere Untersuchung verdienen.
Das Feld hat sich nicht weiterentwickelt, indem es das Manhattan-Diagramm aufgegeben hat. Es hat sich weiterentwickelt, indem es sich geweigert hat, einem Bild mehr Bedeutung zuzumessen, als es verdient.
Häufig gestellte Fragen
Was ist die Hauptbeschränkung eines Manhattan-Plots?
Ein Manhattan-Plot zeigt die Stärke der Assoziation, unterscheidet jedoch nicht von sich aus zwischen echter Biologie und falschen Entdeckungen, ungelöster Linkage-Disequilibrium oder Artefakten der Kohortenstruktur.
Wann ist FDR nützlicher als Bonferroni in GWAS?
FDR ist oft nützlicher in entdeckungsorientierten Arbeitsabläufen, bei denen das Ziel darin besteht, einen breiteren Kandidatensatz für nachgelagerte Feinabstimmungen und funktionale Priorisierungen zu bewahren.
Warum sind lineare Mischmodelle oft besser als PCA allein?
PCA erfasst die Hauptachsen der Abstammung, während LMMs breitere Kovarianz und Verwandtschaft modellieren. In großen oder subtil strukturierten Kohorten führt das oft zu klareren Assoziationsergebnissen.
Wann sollte Fine-Mapping auf eine standardisierte GWAS folgen?
Feinabgleich sollte GWAS folgen, wann immer das Projekt eine kausale Priorisierung benötigt, anstatt nur Spitzenberichte, insbesondere vor funktionaler Validierung oder mechanistischer Nachverfolgung.
Warum GWAS sowohl mit eQTL- als auch mit sQTL-Daten integrieren?
Weil einige Loci hauptsächlich durch Expression wirken, während andere durch Transkriptstruktur oder Isoformnutzung wirken. Die Verwendung beider Ebenen bietet eine umfassendere Sicht auf die regulatorische Funktion.
Ersetzt PRS die Interpretation auf Locus-Ebene?
Nein. PRS fasst verteilte Kohortenebene-Signale zusammen. Es ergänzt das Fine-Mapping und die multi-omische Interpretation, anstatt sie zu ersetzen.
Wie sollte maschinelles Lernen in der GWAS-Forschung eingesetzt werden?
Am besten als Screening-Schicht zur Entdeckung nichtlinearer Interaktionen, nachdem die Studie bereits eine starke Basisassoziation und Strukturkorrektur etabliert hat.
Wie wählt man zwischen PLINK, BOLT-LMM und REGENIE?
Wählen Sie basierend auf der Kohortenarchitektur und dem Workflow-Umfang: PLINK für schnelle Basisregression, BOLT-LMM für große gemischte Modelle menschlicher Kohorten und REGENIE für effiziente großangelegte Hochdurchsatz-Assoziationen.
Referenzen
- Korte A, Farlow A. Die Vorteile und Einschränkungen der Merkmalsanalyse mit GWAS: eine Übersicht. Pflanzenmethoden. 2013;9:29. DOI: 10.1186/1746-4811-9-29
- Marees AT, de Kluiver H, Stringer S, et al. Ein Tutorial zur Durchführung von genomweiten Assoziationsstudien: Qualitätskontrolle und statistische Analyse. International Journal of Methods in Psychiatric Research. 2018;27(2):e1608. DOI: 10.1002/mpr.1608
- Preis AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Hauptkomponentenanalysen korrigieren für Stratifikation in genomweiten Assoziationsstudien. Nature Genetics. 2006;38(8):904-909. DOI: 10.1038/ng1847
- Kang HM, Sul JH, Service SK, et al. Varianzkomponentenmodell zur Berücksichtigung der Stichprobenstruktur in genomweiten Assoziationsstudien. Nature Genetics. 2010;42(4):348-354. DOI: 10.1038/ng.548
- Benner C, Spencer CCA, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: effiziente Variablenauswahl unter Verwendung von Zusammenfassungsdaten aus genomweiten Assoziationsstudien. Bioinformatik. 2016;32(10):1493-1501. DOI: 10.1093/bioinformatics/btw018
- Wang G, Sarkar A, Carbonetto P, Stephens M. Ein einfacher neuer Ansatz zur Variablenauswahl in der Regression, mit Anwendung auf genetische Feinkartierung. Journal of the Royal Statistical Society: Series B (Statistische Methodologie). 2020;82(5):1273-1300. DOI: 10.1111/rssb.12388
- Zhang X, Jiang W, Zhao H. Integration von Expressions-QTLs mit feiner Kartierung über SuSiE. PLoS Genetics. 2024;20(1):e1010929. DOI: 10.1371/journal.pgen.1010929
- Vosa U, Claringbould A, Westra HJ, et al. Großangelegte cis- und trans-eQTL-Analysen identifizieren Tausende von genetischen Loci und polygenen Scores, die die Genexpression im Blut regulieren. Nature Genetics. 2021;53(9):1300-1310. DOI: 10.1038/s41588-021-00913-z
- Li YI, Knowles DA, Humphrey J, et al. Annotation-freie Quantifizierung von RNA-Spleißung mit LeafCutter. Nature Genetics. 2018;50(1):151-158. DOI: 10.1038/s41588-017-0004-9
- Ge T, Chen CY, Ni Y, Feng YCA, Smoller JW. Polygenetische Vorhersage durch Bayessche Regression und kontinuierliche Schrumpfungsprioren. Nature Communications. 2019;10:1776. DOI: 10.1038/s41467-019-09718-5
- Choi SW, Mak TSH, O'Reilly PF. Tutorial: Ein Leitfaden zur Durchführung von Analysen von polygenen Risikoscores. Nature Protocols. 2020;15:2759-2772. DOI: 10.1038/s41596-020-0353-1
- PLINK 2.0 Assoziationsanalyse-Dokumentation. Link: PLINK 2.0 Assoziationsanalyse
- BOLT-LMM Benutzerhandbuch. Link: BOLT-LMM Handbuch
- REGENIE-Dokumentation. Link: REGENIE-Dokumentation
Nur für Forschungszwecke. Nicht für diagnostische Verfahren.
Verwandte Dienstleistungen
- Whole-Genome-Sequenzierung
- Variantenerkennung
- Ganzgenom-SNP-Genotypisierung
- Gezielte Regionssequenzierung
- SNP-Fine-Mapping
- RNA-Seq
- ATAC-Seq
- Vollständige Transkriptsequenzierung (Iso-Seq)
- Multi-Omics-Dienstleistung
- Whole Exome Sequencing - Gesamtes Exom-Sequenzierung
- Human/Maus Whole Exome Sequenzierung
- SNP-Mikroarray
- Genotypisierung durch Sequenzierung (GBS)


