
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Bibliothekskonstruktion für Next-Generation Sequencing (NGS)
Der Aufbau der Bibliothek ist der Schritt im NGS-Workflow, der am anfälligsten für Variabilität ist und die Hauptursache für fehlgeschlagene Sequenzierungsdurchläufe darstellt. Eine gut konstruierte Bibliothek mit optimierten Parametern – Fragmentierungsmethode, molare Verhältnis der Adapter, Protokoll zur Größenselektion und Anzahl der Amplifikationszyklen – kann den Unterschied zwischen einem Projekt ausmachen, das beim ersten Durchlauf veröffentlichungsbereite Daten liefert, und einem, das wiederholte Fehlersuche und Nachsequenzierung erfordert.
Die Kosten einer suboptimalen Bibliothekskonstruktion gehen über die direkten Reagenzkosten hinaus. Eine Bibliothek mit übermäßigem Adapter-Dimer-Gehalt verschwendet Sequenzierungslesungen; eine mit zu vielen PCR-Duplikaten verringert die effektive Abdeckungstiefe; eine falsch größen-selektierte Bibliothek führt zu niedrigeren Mapping-Raten. Wenn sich diese Ineffizienzen in einem Multi-Proben-Projekt summieren, kann der kumulative Datenverlust erheblich sein – vergleichbar mit dem Verlust ganzer Bahnen oder Flusszellen an Sequenzierungskapazität. Bei einem typischen WGS-Projekt mit 96 Proben und 30× Abdeckung bedeutet ein Verlust von 20 % an nutzbaren Lesungen aufgrund von Bibliotheksqualitätsproblemen etwa 0,5-1 Tb an verschwendeten Sequenzierungsdaten und Zehntausende von Dollar an nicht wiederherstellbaren Sequenzierungskosten.
Dieser Leitfaden richtet sich an Forscher, die bereits die grundlegenden Schritte des Bibliotheksbaus verstehen, jedoch quantitative Anleitungen zur Parameteroptimierung benötigen. Er behandelt die wichtigsten einstellbaren Parameter in jedem Schritt des Workflows, die Kompromisse, die mit jeder Wahl verbunden sind, und die QC-Metriken, die eine gute Bibliothek von einer gescheiterten unterscheiden.
Der Fokus ist bewusst praktisch: Nach dem Lesen dieses Leitfadens sollten Sie in der Lage sein, Ihre Wahl der Fragmentierungsmethode zu rechtfertigen, ein angemessenes Verhältnis von Adapter zu Insert-Molar zu berechnen, das richtige Verhältnis der SPRI-Perlen für Ihre Zielfragmentgröße auszuwählen und ein Bioanalyzer-Diagramm zu interpretieren, um festzustellen, ob Ihre Bibliothek die Qualitätskontrolle besteht.
Die in diesem Leitfaden besprochenen Parameter sind Ausgangspunkte – keine festen Regeln. Jedes Labor, jeder Proben-Typ und jede Sequenzierungsplattform kann geringfügige Anpassungen dieser empfohlenen Werte erfordern. Der effektivste Ansatz besteht darin, eine Basislinie mit den empfohlenen Parametern festzulegen und dann wichtige Variablen (PCR-Zyklen, Bead-Verhältnisse) durch kleinmaßstäbliche Pilotversuche zu titrieren, um die optimalen Einstellungen für Ihren spezifischen Arbeitsablauf und Proben-Typ zu bestimmen.
Der Kernarbeitsablauf auf einen Blick
Jeder Illumina-Bibliothekskonstruktionsworkflow folgt der gleichen Abfolge von Schritten, unabhängig vom spezifischen Kit, Protokoll oder dem Ausgangsmaterialtyp:
- FragmentierungDNA wird in Fragmente der Zielgröße zerbrochen.
- Endreparatur und A-TailingFragmentenden sind abgestumpft, phosphoryliert und mit einem A-Schwanz versehen.
- Adapter-LigationSequenzierungsadapter werden an die Fragmente ligiert.
- Bereinigung und GrößenwahlUninkorporierte Adapter und Fragmente außerhalb des Zielbereichs werden mit SPRI-Magnetperlen entfernt.
- BibliotheksverstärkungDie PCR-Amplifikation erhöht den Bibliotheksausstoß (PCR-freie Methoden überspringen diesen Schritt).
- Bibliotheks-QCDie endgültige Bibliothek wird quantifiziert und qualitätsgeprüft.
Die Kernschritte des Illumina-Bibliothekskonstruktions-Workflows – Fragmentierung, Endreparatur, Adapterligierung, Reinigung, Amplifikation und Qualitätskontrolle – können als linearer Prozess visualisiert werden. Die Eingangs-DNA tritt links ein, durchläuft jeden Transformationsschritt und verlässt rechts die Anlage als sequenzierbare Bibliothek. Die Qualität der endgültigen Bibliothek hängt von den kumulativen Auswirkungen der Parameterentscheidungen in jedem Schritt ab, wobei frühere Schritte den größten Einfluss haben, da Fehler im Workflow vorwärts propagiert werden.
Jeder dieser Schritte hat anpassbare Parameter, die die endgültige Qualität der Bibliothek erheblich beeinflussen. Die folgenden Abschnitte bieten quantitative Hinweise für jede Parameterwahl.
 Abbildung 1. Sechs-Schritte-Illumina-Bibliothekskonstruktionsworkflow — von der DNA-Fragmentierung bis zur finalen Qualitätskontrolle
Abbildung 1. Sechs-Schritte-Illumina-Bibliothekskonstruktionsworkflow — von der DNA-Fragmentierung bis zur finalen Qualitätskontrolle
Bildunterschrift: Der sechs Schritte umfassende Illumina-Bibliothekskonstruktionsworkflow, der den linearen Verlauf von der Fragmentierung über die Endreparatur, Adapterligatur, Größenauswahl, Amplifikation bis zur finalen Qualitätskontrolle zeigt, wobei frühe Fehler sich im Verlauf des Prozesses fortpflanzen.
Fragmentierung — Vergleich von mechanischen, enzymatischen und Tagmentationsansätzen
Fragmentierung ist der erste variable Parameter im Workflow zur Bibliothekskonstruktion und hat einen messbaren Einfluss auf die Datenqualität im Nachgang. Es stehen drei Ansätze zur Verfügung, die jeweils unterschiedliche Eigenschaften aufweisen, die die Abdeckungsuniformität, Reproduzierbarkeit und Eingangsflexibilität beeinflussen.
Mechanische Fragmentierung verwendet akustische Energie (Covaris) oder Nebelung, um DNA in Fragmente zu zerbrechen. Der Prozess ist rein physikalisch – es sind keine Enzyme beteiligt – was bedeutet, dass er im Wesentlichen keine sequenzabhängige Verzerrung erzeugt. Dies macht die mechanische Fragmentierung zur bevorzugten Methode für WGS-Projekte, bei denen eine einheitliche Abdeckung aller genomischen Regionen, einschließlich hoch-GC-promotoren und niedrig-GC-Introns, von entscheidender Bedeutung ist. Die Nachteile sind höhere Kosten für das Instrument, längere Bearbeitungszeiten und eine geringere Durchsatzrate im Vergleich zu enzymatischen Methoden.
Enzymatische Fragmentierung verwendet einen Nuclease-Cocktail (typischerweise NEBNext Ultra II oder KAPA HyperPlus), um Doppelstrangbrüche einzuführen. Die enzymatische Fragmentierung ist skalierbarer als mechanisches Zerschneiden und funktioniert über einen breiteren Eingangsbereich, von sub-Nanogramm-Mengen bis zu mehreren Mikrogramm. Das Bias-Profil hängt von der Enzymformulierung ab – einige Enzymmischungen erzeugen eine nahezu mechanische Qualität mit signifikant höherem Durchsatz.
Tagmentierung (auf Tn5-basierend) verwendet eine hyperaktive Tn5-Transposase, die DNA gleichzeitig fragmentiert und Adaptersequenzen in einer einzigen Reaktion einfügt. Dies reduziert die praktische Arbeitszeit von etwa 30 Minuten auf unter 5 Minuten und funktioniert mit so wenig wie 1 ng Eingangs-DNA. Der Nachteil ist, dass Tn5 in Regionen mit niedrigem GC-Gehalt messbare Verzerrungen zeigt, wo die Abdeckung auf 60-70 % des genomischen Durchschnitts sinken kann. Die Tagmentierung eignet sich am besten für Anwendungen, bei denen Geschwindigkeit und niedrige Eingangsmenge über absolute Abdeckungsuniformität priorisiert werden.
| Parameter | Mechanisch (Covaris) | Enzymatisch | Tagmentierung (Tn5) |
|---|---|---|---|
| DNA-Bereich eingeben | 50 ng – 5 µg | 0,5 ng – 1 µg | 1–50 ng |
| GC-Bias | Niedrigste | Niedrig bis mäßig | Moderat (niedrig-GC-Regionen) |
| Reproduzierbarkeit (CV) | <10% | 10–20 % | 15–25 % |
| Praktische Zeit | 10–15 Minuten (plus Instrument) | 5–10 Minuten | 2–5 Minuten |
| Am besten geeignet für | WGS, hochwertige Proben | Allgemeiner Zweck, FFPE, cfDNA | Niedriger Input, schnelle Screening, bakterielle Genome |
AuswahllogikFür WGS-Projekte, bei denen eine einheitliche Abdeckung über den GC-Gehalt entscheidend ist, erzeugt mechanische Fragmentierung die geringste Verzerrung. Für allgemeine Arbeitsabläufe, die verschiedene Probenarten verarbeiten, bietet die enzymatische Fragmentierung das beste Gleichgewicht zwischen Bequemlichkeit und Qualität. Tagmentation ist ideal für Anwendungen mit niedrigem Input oder hohem Durchsatz, bei denen Geschwindigkeit über die Gleichmäßigkeit der Abdeckung priorisiert wird.
Bei der Auswahl einer Fragmentierungsmethode bestimmt die nachgelagerte Anwendung die geeignete Wahl. Zum Beispiel, RNA-Sequenzierungsprojekte Verwenden Sie die wärmeinduzierte Fragmentierung von cDNA anstelle einer der drei DNA-Fragmentierungsmethoden, wodurch der Fragmentierungsschritt grundlegend anders ist als bei DNA-Bibliotheks-Workflows.
FragmentierungsqualitätsprüfungBevor Sie mit der Endreparatur und Ligation fortfahren, ist es ratsam, die Fragmentgrößenverteilung mit einem Bioanalyzer oder TapeStation zu überprüfen. Der Verlauf sollte eine unimodale Verteilung zeigen, die auf der Zielgröße zentriert ist. Eine bimodale Verteilung oder ein breiter Peak, der >500 bp umfasst, weist auf suboptimale Fragmentierungsbedingungen hin und kann eine Anpassung der Scherzeit oder der Enzymkonzentration erforderlich machen, bevor Sie mit dem restlichen Workflow fortfahren.
Forscher, die groß angelegte Projekte planen, können nutzen Dienstleistungen zur gesamten Genomsequenzierung wo Fragmentierung und Bibliothekskonstruktion für jeden Proben-Typ optimiert sind.
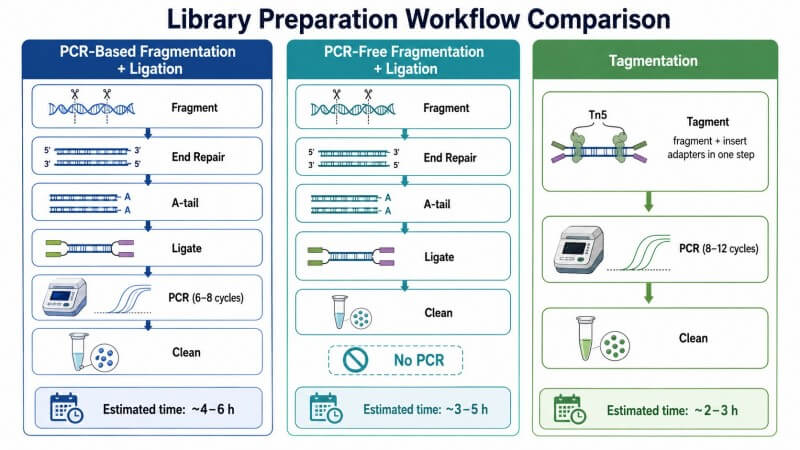 Abbildung 2. Vergleich der Fragmentierungsmethoden – Abdeckungsbias-Profile über den GC-Gehalt für mechanische, enzymatische und Tagmentationsansätze
Abbildung 2. Vergleich der Fragmentierungsmethoden – Abdeckungsbias-Profile über den GC-Gehalt für mechanische, enzymatische und Tagmentationsansätze
Bildunterschrift: Vergleichende Abdeckungsbias-Profile über den GC-Gehalt für drei Fragmentierungsmethoden, die zeigen, dass mechanische Fragmentierung die gleichmäßigste Abdeckung erzeugt, enzymatische Fragmentierung mit niedrigem bis moderatem Bias und Tagmentierung mit messbarem Bias in Regionen mit niedrigem GC-Gehalt.
Adapter-Ligation — Molarverhältnis-Optimierung
Das molare Verhältnis von Adaptern zu Insert-DNA-Fragmenten ist eines der am häufigsten falsch optimierten Parameter beim Bibliotheksaufbau. Wenn es falsch ist, produziert man entweder Adapter-Dimere (zu viel Adapter) oder ineffizient markierte Bibliotheken (zu wenig Adapter). Das optimale Verhältnis hängt von der Konzentration der ligierbaren Fragmentenden ab, die sowohl durch die Masse der Eingangs-DNA als auch durch die durchschnittliche Fragmentgröße bestimmt wird. Eine gegebene Masse an kurzen Fragmenten hat pro Masseneinheit mehr Enden als die gleiche Masse an langen Fragmenten, daher muss das molare Verhältnis die Anzahl der verfügbaren Enden berücksichtigen.
Für den Standardaufbau von Illumina-Bibliotheken liegt das empfohlene Verhältnis von Adapter zu Insert im Bereich von 10:1 bis 50:1, abhängig von der Eingabemenge:
- >500 ng Eingabe → 10:1 bis 20:1 Verhältnis (ausreichende Ligationseffizienz, minimales Dimer-Risiko)
- 100–500 ng Eingabe → Verhältnis von 20:1 bis 30:1
- 10–100 ng Eingabe → 30:1 bis 50:1 Verhältnis (höheres Verhältnis erforderlich, um die niedrigere Kollisionswahrscheinlichkeit auszugleichen)
- <10 ng Eingabe → 50:1 bis 100:1 Verhältnis; Adapter-Dimer wird zu einem signifikanten Risiko, und spezialisierte Low-Input-Kits mit Stem-Loop-Adaptern werden gegenüber Standard-Y-Adaptern empfohlen.
Y-Adapter vs. Stem-Loop-AdapterStandard-Y-Adapter haben zwei nicht-komplementäre Arme, die eine gabelartige Struktur bilden. Während der Ligation können beide Arme an Ligationsevents teilnehmen, und wenn kein Insert vorhanden ist, können sich die beiden Arme gegenseitig ligieren und Adapter-Dimere bilden. Stem-Loop-Adapter verwenden eine Haarnadelstruktur, die die Adapter-zu-Adapter-Ligation verhindert und die Dimmerbildung im Vergleich zu Y-Adaptern um 50-80 % reduziert. Für die Konstruktion von Low-Input-Bibliotheken, bei denen Adapter-Dimere der primäre Fehlerursache sind, bieten Stem-Loop-Adapter eine signifikante Qualitätsverbesserung.
Adapter-Dimer-Kontamination ist auf einem Bioanalyzer-Trace als ein Peak bei 120–140 bp nachweisbar. Bei Werten unter 5 % der gesamten Bibliotheksmasse haben Dimeren minimale Auswirkungen auf die Sequenzierung; über 5 % verbrauchen sie Sequenzierungslesungen, ohne verwertbare Daten zu erzeugen, was effektiv den nutzbaren Datenoutput des Projekts reduziert. Bei Werten über 20 % werden Adapter-Dimeren zur dominierenden Spezies in der Bibliothek, und der Sequenzierungslauf wird überwiegend unalignierbare Lesungen produzieren.
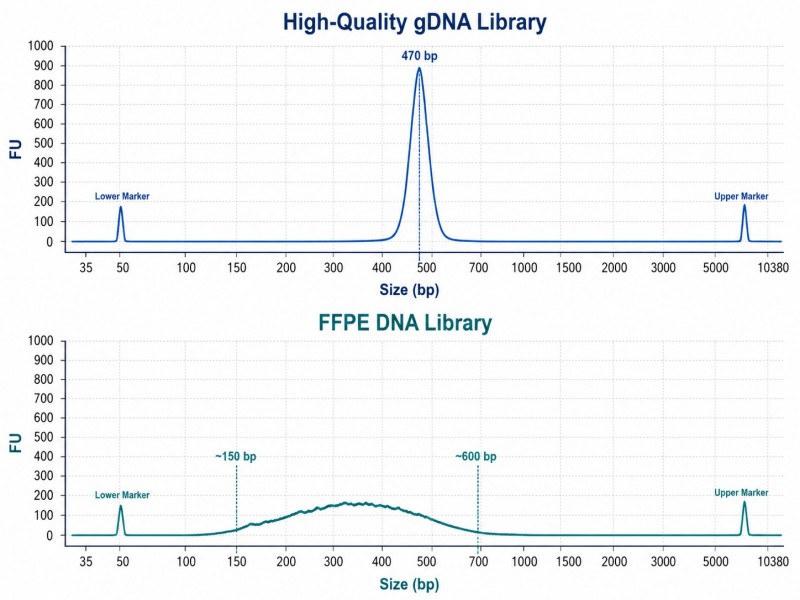 Abbildung 3. Verhältnis von Adapter-Molarität zu Bibliotheksausbeute und Dimerbildung — quantitative Beziehung
Abbildung 3. Verhältnis von Adapter-Molarität zu Bibliotheksausbeute und Dimerbildung — quantitative Beziehung
Beschriftung: Quantitative Beziehung zwischen dem molaren Verhältnis von Adapter zu Insert und dem Bibliotheksausstoß (blau) sowie der Bildung von Adapter-Dimeren (rot), die das optimale Betriebsfenster zwischen 10:1 und 50:1 je nach eingegebener DNA-Menge zeigt.
Endreparatur und A-Tailing — Ein kritischer Zwischenschritt
Nach der Fragmentierung haben DNA-Fragmente heterogene Enden – einige haben 5'- oder 3'-Überhänge, einige sind stumpf und einige können beschädigte oder modifizierte Enden aufweisen. Der Schritt zur Endreparatur verwendet eine Kombination von Enzymen – typischerweise T4-DNA-Polymerase (füllt 5'-Überhänge auf und entfernt 3'-Überhänge) und T4-Polynukleotidkinase (phosphoryliert 5'-Enden) – um einheitliche, stumpf-endige, 5'-phosphorylierte Fragmente zu erzeugen.
A-Tailing folgt unmittelbar: Eine Taq-Polymerase fügt jedem stumpfen Fragment ein einzelnes Adenosin an das 3'-Ende hinzu. Dieser A-Überhang ist komplementär zum einzelnen T-Überhang an den standardmäßigen Illumina-Adaptern, was eine effiziente Adapterligatur ermöglicht und gleichzeitig verhindert, dass die Adapter miteinander ligieren.
Der entscheidende Parameter bei der Endreparatur ist das Verhältnis von Enzym zu DNA. Ein unzureichendes Enzym lässt einen Teil der Enden unrepariert oder ohne A-Schwanz, was die Anzahl der Fragmente verringert, die erfolgreich an Adapter ligieren können. Dies führt zu einer Bibliothek mit einem niedrigeren Endertrag als erwartet aus der Menge an Eingangs-DNA. Ein häufiges Symptom ist eine Bibliothek, die die Qubit-Quantifizierung besteht, aber eine niedrige Clusterdichte auf der Flusszelle zeigt – was darauf hindeutet, dass viele der quantifizierten Moleküle funktionale Adaptersequenzen an beiden Enden fehlen.
Die meisten kommerziellen Bibliothekskonstruktion-Kits enthalten optimierte Enzymmischungen, die diese Aktivitäten ausbalancieren. Bei Verwendung eines benutzerdefinierten Protokolls ist eine 30-minütige Endreparatur bei 20 °C, gefolgt von einer 30-minütigen A-Tailing bei 37 °C, ein standardmäßiger Ausgangspunkt, der je nach spezifischer Enzymformulierung angepasst werden kann.
Auswirkungen der A-Tailing-Effizienz auf die nachgelagerte AdapterligaturDer A-Tailing-Schritt wird manchmal als routinemäßige enzymatische Reaktion betrachtet, aber seine Effizienz bestimmt direkt den maximal möglichen Ligationsertrag. Wenn nur 80 % der Fragmente einen A-Überhang erhalten, sinkt die theoretische maximale Ligationseffizienz von 50 % (wenn jedes Fragmentpaar A-tailed ist) auf etwa 40 %. Diese 20 % Reduktion der effektiven Moleküle führt direkt zu einem niedrigeren endgültigen Bibliotheksausbeute. Bei Proben mit niedrigem Input, bei denen jedes Molekül zählt, ist die Gewährleistung eines vollständigen A-Tailings eine der einfachsten Möglichkeiten, die Bibliotheksausbeute ohne zusätzliche PCR-Zyklen zu maximieren. Genomdatenanalyse Der downstream-Bereich ist ebenfalls betroffen, da Bibliotheken mit suboptimalem Ertrag möglicherweise nicht genügend Abdeckung für eine zuverlässige Variantenentdeckung bieten.
Größenauswahl — Ein praktischer Leitfaden für das Verhältnis von SPRI-Perlen
SPRI (Solid Phase Reversible Immobilization) bead-basierte Größenselektion ist die am häufigsten verwendete Methode zur Entfernung unerwünschter Fragmentgrößen aus NGS-Bibliotheken. Der entscheidende Parameter ist das Verhältnis von Bead- zu Probenvolumen, das den Größenbereich der zurückgehaltenen Fragmente bestimmt. Das Prinzip ist einfach: Bei höheren Bead-Verhältnissen werden kleinere Fragmente erfasst; bei niedrigeren Bead-Verhältnissen binden nur größere Fragmente. Durch die Durchführung von zwei aufeinanderfolgenden Selektionen kann ein präzises Zielgrößenfenster isoliert werden.
Verstehen der Beziehung des PerlenverhältnissesSPRI-Perlen funktionieren, indem sie DNA-Fragmente in Anwesenheit eines Crowdings-Agenten (PEG) binden. Die Bindungseffizienz ist größenabhängig: Bei einer bestimmten PEG-Konzentration binden größere Fragmente bevorzugt, während kleinere Fragmente in Lösung bleiben. Ein Verhältnis von 0,6× bedeutet, dass das Volumen der Perlen 60 % des Probenvolumens beträgt; bei diesem Verhältnis binden Fragmente über etwa 500–600 bp an die Perlen und können erfasst werden. Der Überstand enthält Fragmente unterhalb dieser Schwelle, einschließlich der meisten Adapter-Dimere. Ein Verhältnis von 0,8× erfasst Fragmente über etwa 200–300 bp. Der Unterschied zwischen zwei aufeinanderfolgenden Verhältnissen definiert das Größenfenster.
| Zielfragmentbereich | Erster Perlenverhältnis (Rechte Seite) | Zweites Perlenverhältnis (Linke Seite) | Anwendung |
|---|---|---|---|
| 200–300 bp | 0,6 × (Perlen wegwerfen) | 0,8 × (Perlen behalten) | Amplicon-, cfDNA-Bibliotheken |
| 300–500 bp | 0,6 × (Perlen wegwerfen) | 0,7 × (Perlen behalten) | Standard WGS-, WES-Bibliotheken |
| 500–800 bp | 0,5× (Perlen entsorgen) | 0,6 × (Perlen behalten) | Long-Insert-Bibliotheken, Mate-Paar |
Wie die Auswahl der doppelseitigen Größe funktioniertDie erste (rechte) Auswahl verwendet ein niedriges Perlenverhältnis, um große Fragmente und lange Adapter-Dimere zu binden, die mit den Perlen verworfen werden. Der Überstand, der die Fragmente der Zielgröße enthält, wird dann in ein zweites Röhrchen mit einem höheren Perlenverhältnis übertragen, um die gewünschten Fragmente zu erfassen. Der verbleibende Überstand mit kurzen Fragmenten und Adapter-Dimeren wird verworfen.
Die Präzision der Größenauswahl hängt von der Differenz zwischen den beiden Verhältnissen ab. Eine größere Spanne (z. B. 0,5× bis 0,8×) behält einen breiteren Größenbereich bei; eine engere Spanne (z. B. 0,6× bis 0,7×) führt zu einer engeren Verteilung, jedoch mit einem niedrigeren Gesamtertrag. Für die meisten WGS-Anwendungen bietet eine doppelseitige Auswahl von 0,6×/0,7× ein gutes Gleichgewicht zwischen Ertrag und Präzision. Für Anwendungen, die eine genauere Größenkontrolle erfordern – wie z. B. den Aufbau von cfDNA-Bibliotheken, bei denen die Zielfragmente bereits kurz sind – ist eine engere Spanne zwischen den beiden Verhältnissen angemessen.
Einseitige vs. beidseitige ReinigungEine einseitige Reinigung (Verhältnis von einem Bead, ein Magnet-Schritt) entfernt nur Fragmente unterhalb eines Schwellenwerts. Sie ist schneller, entfernt jedoch keine großen Fragmente, die die Clusterqualität auf dem Flusszellen beeinträchtigen können. Für die meisten Protokolle zur Bibliothekskonstruktion wird eine doppelseitige Reinigung empfohlen, um sicherzustellen, dass sowohl große als auch kleine Fragmente entfernt werden.
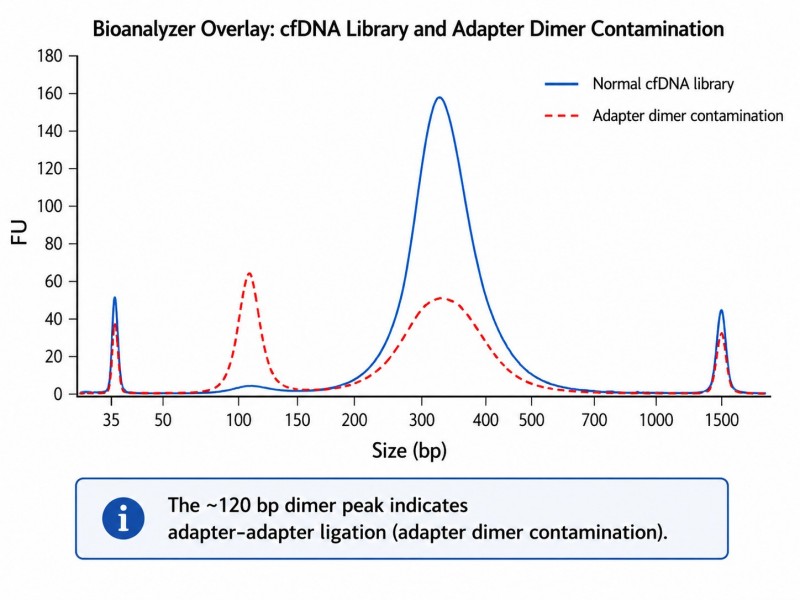 Abbildung 4. SPRI-Perlenverhältnis-Leitfaden — doppelseitige Größenwahlbereiche für verschiedene Zielfragmentgrößen
Abbildung 4. SPRI-Perlenverhältnis-Leitfaden — doppelseitige Größenwahlbereiche für verschiedene Zielfragmentgrößen
Bildunterschrift: SPRI-Perlenverhältnis-Auswahlleitfaden, der das doppelseitige Größenwahlprotokoll für drei Zielfragmentbereiche – 200-300 bp, 300-500 bp und 500-800 bp – mit entsprechenden Perlenverhältnis-Paaren und Anwendungszusammenhängen zeigt.
PCR-Amplifikation — Zyklusnummer vs. Duplikationsrate
Die PCR-Amplifikation ist für die meisten Bibliothekskonstruktions-Workflows notwendig, aber jeder zusätzliche Zyklus erhöht die Duplikationsrate und führt zu Verzerrungen. Die optimale Zyklusanzahl hängt hauptsächlich von der Menge der Eingangs-DNA ab.
Die Beziehung zwischen der Zählung der Zyklen und der Duplikation ist nicht linear. In den ersten 4–6 Zyklen befindet sich die Amplifikation in der exponentiellen Phase, in der jedes Template-Molekül ein einzigartiges Tochtermolekül produziert und die Duplikation minimal ist. Nach 8–10 Zyklen beginnt die Reaktion zu sättigen: amplifizierte Moleküle beginnen, sich wieder anzulagern und PCR-Duplikate zu bilden, und die effektive Rate der Erzeugung neuer einzigartiger Moleküle verlangsamt sich. Über 10 Zyklen hinaus fügt jeder zusätzliche Zyklus etwa 5–10% mehr Duplikate hinzu, ohne die einzigartige Komplexität der Bibliothek proportional zu erhöhen.
Eine hochgenaue Polymerase (Fehlerquote <10⁻⁶ pro Base) ist entscheidend, um die Einführung von artefaktischen Mutationen während der Bibliothekskonstruktion zu minimieren. Die Standard-Taq-Polymerase führt Fehler mit einer Rate von etwa 10⁻⁴ bis 10⁻⁵ pro Base ein, was in sensiblen Anwendungen wie der Erkennung seltener Varianten oder der Einzelzell-Sequenzierung zu falsch-positiven Variantenaufrufen führen kann. KAPA HiFi, Q5 und Pfu-basierte Polymerasen werden häufig in der NGS-Bibliothekskonstruktion verwendet, da sie ein ausgewogenes Verhältnis von Ausbeute, Genauigkeit und Amplifikationsbias bieten.
| Eingangs-DNA | Empfohlene PCR-Zyklen | Erwartete Duplikationsrate |
|---|---|---|
| >1 µg (für PCR-frei) | 0 (PCR-frei) | <5% |
| 100 ng–1 µg | 4–8 | 5–15 % |
| 10–100 ng | 8–10 | 10–20 % |
| 1–10 ng | 10–12 | 15–30 % |
| <1 ng | 12–14 | 20–50 % |
Für Anwendungen, bei denen die Duplikationsrate entscheidend ist (WGS-Variantenerkennung, Erkennung seltener Varianten), ist der PCR-freie Bibliotheksaufbau der bevorzugte Ansatz, wann immer die Eingabemenge dies zulässt. Für Proben mit niedrigem Input, bei denen PCR-frei nicht möglich ist, ist die Verwendung einer hochfidelitäts Polymerase und die Begrenzung der Zyklusanzahl auf das Minimum, das erforderlich ist, um einen ausreichenden Bibliotheksausbeute zu erzielen, die beste praktische Strategie.
Für Projekte mit spezifischen Abdeckungsanforderungen, gezielte Sequenzierungsdienste kann helfen, die Parameter für den Bibliotheksaufbau für die Zielregion von Interesse zu optimieren.
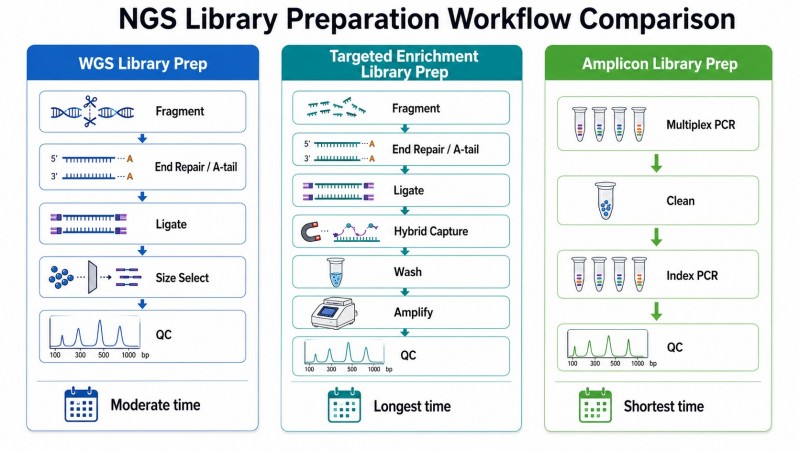 Abbildung 5. PCR-Zyklusanzahl vs. Duplikationsrate — quantitative Beziehung über Eingangs-DNA-Niveaus
Abbildung 5. PCR-Zyklusanzahl vs. Duplikationsrate — quantitative Beziehung über Eingangs-DNA-Niveaus
Bildunterschrift: Quantitative Beziehung zwischen der PCR-Zyklenanzahl und der Duplikationsrate bei verschiedenen Eingangs-DNA-Niveaus, die den Übergang von der exponentiellen zur Sättigungsphase und den optimalen Zyklusbereich für jede Eingabekategorie zeigt.
Alles zusammenfügen — Ein Beispiel für Projektplanung
Um zu veranschaulichen, wie diese Parameter in der Praxis interagieren, betrachten wir einen Forscher, der ein Sequenzierungsprojekt des gesamten menschlichen Genoms mit 48 Proben plant. Die Proben bestehen aus 40 hochwertigen Blut-DNA-Proben (Eingang >1 µg jeweils) und 8 FFPE-Tumorproben (Eingang ~100 ng jeweils, degradiert). Ein einzelnes Protokoll zur Bibliothekskonstruktion wird nicht optimal für beide Probenarten geeignet sein.
Für die 40 Blut-DNA-Proben ist der PCR-freie Bibliotheksaufbau mit mechanischer Fragmentierung und einer doppelseitigen SPRI-Reinigung von 0,6×/0,7× der bevorzugte Ansatz. Die PCR-freie Methode beseitigt Duplikationsbias, und die mechanische Fragmentierung sorgt für eine gleichmäßige GC-Abdeckung. Die Ziel-Insertgröße beträgt 350-500 bp, und die endgültigen Bibliotheken sollen >5 nM durch qPCR ergeben.
Für die 8 FFPE-Proben ist eine PCR-basierte Bibliothekskonstruktion mit enzymatischer Fragmentierung (die einen Schritt zur Reparatur von DNA-Schäden umfasst) erforderlich. Das Verhältnis von Adaptern zu Inserts sollte auf 30:1 erhöht werden, um den geringeren effektiven Input aus degradierter DNA auszugleichen. Die PCR-Zyklen sollten auf 8-10 begrenzt werden, um die Amplifikation von Artefakten zu vermeiden. Das Größenwahlfenster sollte verbreitert werden (0,6×/0,8×), um die breitere Fragmentgrößenverteilung, die für FFPE-Proben typisch ist, zu berücksichtigen.
Die beiden Bibliothekssätze können nicht im selben Flusszellenbereich zusammengeführt werden, ohne die Ladekonzentrationen anzupassen, da die PCR-freien Bibliotheken eine erheblich höhere Molarität aufweisen werden. Eine separate Quantifizierung durch qPCR für jeden Satz, gefolgt von einer äquimolaren Zusammenführung innerhalb jedes Satzes, gewährleistet eine konsistente Clusterdichte über alle Proben hinweg.
Dieses Beispiel zeigt, warum die in diesem Leitfaden besprochenen Parameter auf einer pro Probenart-Basis angewendet werden müssen und nicht als universelles Protokoll. NGS-Sequenzierungsdienste Mit Fachkenntnissen in der Erstellung anwendungsspezifischer Bibliotheken können diese variablen Parametersätze über verschiedene Probenarten innerhalb eines einzigen Projekts verwaltet werden.
Anwendungsspezifische Bibliothekskonstruktion — WGS vs. RNA vs. Epigenomik
Die optimalen Parameter für den Bibliotheksaufbau unterscheiden sich erheblich je nach nachgelagerter Anwendung:
| Anwendung | Fragmentierung | Adapter | PCR | Schlüssel-QC |
|---|---|---|---|---|
| Whole-Genome-Sequenzierung | Mechanisch oder enzymatisch | Standard-Y-Adapter | PCR-frei oder 4–8 Zyklen | Duplikationsrate <15% |
| RNA-Seq (mRNA) | Wärmeinduzierte Fragmentierung von cDNA | RNA-spezifische Adapter | 10–14 Zyklen | RIN ≥ 7, rRNA-Depletionseffizienz |
| Epigenomik (WGBS) | Enzymatisch (bisulfitkompatibel) | Methylierte Adapter | 8–12 Zyklen | Bisulfite-Konversionsrate >99% |
| ChIP-seq | Enzymatische oder Tagmentation | Standard-Y-Adapter | 10–14 Zyklen | Fragmente, die in der erwarteten Größe angereichert sind |
| Metagenomik (Shotgun) | Enzymatisch (niedriger Input) | Standard-Y-Adapter | 8–12 Zyklen | Keine Kontamination mit Wirts-DNA |
Die wichtigste Erkenntnis ist, dass es keine "Einheitsgröße für alle" Bibliothekskonstruktionsparameter gibt. Ein für WGS optimiertes Protokoll wird bei cfDNA oder RNA-seq aufgrund von Unterschieden in der Eingabemenge, der Fragmentgrößenverteilung und der Adapterkompatibilität schlecht abschneiden.
Praktisches Beispiel — WES-Bibliothekskonstruktion vs. WGSDie Ganzexom-Sequenzierung erfordert einen zusätzlichen Hybridisierungsfangschritt nach der initialen Bibliothekskonstruktion. Die initiale Bibliothek muss die gleichen Qualitätskontrollstandards wie eine WGS-Bibliothek erfüllen, jedoch mit einer zusätzlichen Einschränkung: Die Bibliothek muss eine ausreichende Komplexität aufweisen, um den Fangschritt ohne übermäßige Duplikatraten zu überstehen. Das bedeutet, dass die Konstruktion der WES-Bibliothek weniger PCR-Zyklen als eine WGS-Bibliothek mit demselben Input verwenden sollte, da der Fangschritt eine zusätzliche Amplifikation einführt, die die Duplikationsrate erhöht.
Praktisches Beispiel — cfDNA-BibliothekskonstruktionDie Konstruktion von cfDNA-Bibliotheken ist arguably am empfindlichsten gegenüber der Optimierung von Parametern. Die Eingabemenge (1–50 ng) und die Fragmentgröße (~167 bp) liegen beide außerhalb des optimalen Bereichs für Standardprotokolle. Das Verhältnis von Adapter zu Insert muss sorgfältig kontrolliert werden: Zu niedrig, und die Ligationseffizienz leidet; zu hoch, und die Kontamination mit Adapter-Dimeren wird schwerwiegend, da die kurzen cfDNA-Fragmente durch Größenauswahl nicht effektiv von Dimeren getrennt werden können. Für cfDNA-Workflows werden spezialisierte Kits zur Konstruktion von Bibliotheken mit niedrigem Input, die Stem-Loop-Adapter und optimierte Beadraten enthalten, empfohlen.
NGS-Sequenzierungsdienste Mit anwendungsspezifischen Bibliothekskonstruktionsprotokollen können die Parametersätze an die Anforderungen jedes Projekts angepasst werden.
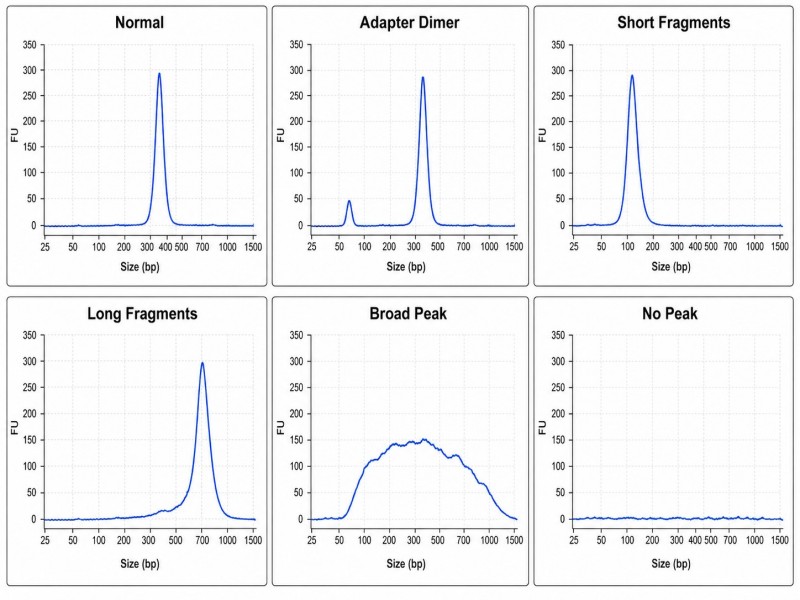 Abbildung 6. Anwendungspezifische Bibliothekskonstruktion — Parameterunterschiede bei fünf gängigen NGS-Anwendungen
Abbildung 6. Anwendungspezifische Bibliothekskonstruktion — Parameterunterschiede bei fünf gängigen NGS-Anwendungen
Überschrift: Vergleichstabelle der Parameter für fünf NGS-Anwendungen—WGS, RNA-seq, WGBS, ChIP-seq und Metagenomik—die Unterschiede in der Fragmentierungsmethode, dem Adaptertyp, der Anzahl der PCR-Zyklen und den wichtigsten QC-Metriken für jede Anwendung zeigt.
Bibliotheks-QC — Quantifizierbare Akzeptanzkriterien
Ein systematischer QC-Workflow mit vordefinierten Pass/Fail-Schwellenwerten verhindert, dass minderwertige Bibliotheken Sequenzierungskapazitäten verschwenden. Die folgenden Kriterien stellen praktische Standards für die meisten Illumina-Bibliothekskonstruktionsprojekte dar. Diese Schwellenwerte basieren auf empirischen Erfahrungen aus Tausenden von Bibliotheken und spiegeln die Leistung wider, die erforderlich ist, um hochwertige Sequenzierungsdaten auf modernen Illumina-Plattformen zu erzeugen.
| QC-Metrik | Methode | Pass | Vorsicht | Fehler |
|---|---|---|---|---|
| Bibliothekskonzentration | qPCR | >2 nM | 1–2 nM | <1 nM |
| Adapter-Dimer-Inhalt | Bioanalysator | <5% der Gesamtmasse | 5–10 % | >10% |
| Durchschnittliche Fragmentgröße | Bioanalysator | Innerhalb von ±10 % des Ziels | ±10–20 % | ±20% |
| Duplizierungsrate (WGS) | Berechnend | <15% | 15–30 % | >30% |
| qPCR vs. Qubit-Verhältnis | Beide | 0,5–2,0 | 2,0–3,0 | >3.0 |
Interpretation der kombinierten QC-ErgebnisseKein einzelner Maßstab ist ausreichend, um die Qualität einer Bibliothek zu bewerten. Eine Bibliothek mit mehr als 10 % Adapterdimer-Inhalt kann die Sequenzierungs-QC bestehen, wenn die Gesamtkonzentration hoch ist, aber sie wird einen Teil der Reads auf nicht informative Sequenzen verschwenden. Eine Bibliothek mit niedriger Konzentration (<1 nM) kann zwar auf die Flusszelle geladen werden, wird jedoch wahrscheinlich eine niedrige Clusterdichte erzeugen und die Kapazität der Flusszelle nicht optimal nutzen. Der zuverlässigste Indikator für eine gute Bibliothek ist eine, die alle QC-Schwellenwerte gleichzeitig besteht.
EnzymbatchverfolgungEin oft übersehener Faktor bei der Reproduzierbarkeit von Bibliothekskonstruktionen ist die Variabilität zwischen Enzymchargen. Enzyme, die bei Fragmentierung, Endreparatur, Ligation und Amplifikation verwendet werden, sind biologische Reagenzien mit inhärenter Chargen-zu-Chargen-Variabilität. Für Projekte, die sich über mehrere Chargen erstrecken, ist es eine gute Praxis, eine kleine Aliquote jeder Enzymcharge für einen Vergleich nebeneinander aufzubewahren. Eine Kontrollprobe mit bekannter Leistung sollte beim Wechsel zu einer neuen Charge einbezogen werden, um konsistente Ergebnisse zu validieren.
Das Verhältnis von qPCR zu Qubit ist ein besonders nützliches Diagnosewerkzeug. qPCR misst nur amplifizierbare, adapter-ligierte Bibliotheksmoleküle, während Qubit gesamtes doppelsträngiges DNA im Sample misst. Wenn Qubit signifikant höher liest als qPCR (Verhältnis >3), enthält die Bibliothek wahrscheinlich einen hohen Anteil an nicht amplifizierbarer DNA – typischerweise Adapter-Dimere, unligierte Fragmente oder Primer-Artefakte, die keine Sequenzierungsdaten erzeugen werden. Ein Verhältnis nahe 1 weist auf eine saubere Bibliothek hin, bei der die meisten gemessenen DNA-Moleküle funktionale Bibliotheksmoleküle sind.
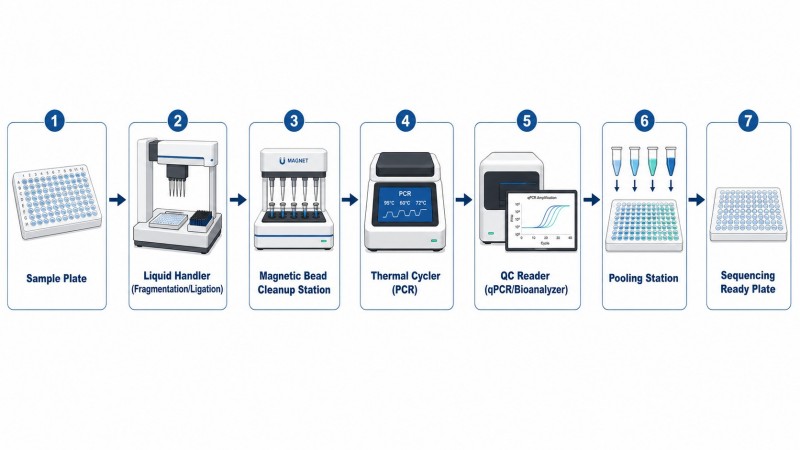 Abbildung 7. Entscheidungsflussdiagramm zur Qualitätskontrolle der Bibliothek – Bestehen/Nichtbestehen-Kriterien für jeden QC-Prüfpunkten
Abbildung 7. Entscheidungsflussdiagramm zur Qualitätskontrolle der Bibliothek – Bestehen/Nichtbestehen-Kriterien für jeden QC-Prüfpunkten
Bildunterschrift: Flussdiagramm zur Entscheidung der Bibliotheks-QC, das die Pass-/Vorsicht-/Fehlergrenzen für fünf wichtige Kennzahlen zeigt – qPCR-Konzentration, Adapter-Dimer-Gehalt, Fragmentgröße, Duplikationsrate und qPCR-zu-Qubit-Verhältnis – mit empfohlenen Maßnahmen an jedem Entscheidungspunkt.
Wie CD Genomics die Bibliothekskonstruktion unterstützt
CD Genomics bietet umfassende Dienstleistungen zur Bibliothekskonstruktion an, die das gesamte Spektrum der Illumina-kompatiblen Methoden und Probenarten abdecken.
Verfügbare MethodenUnser Labor führt mechanische Fragmentierung, enzymatische Fragmentierung und tagmentationsbasierte Bibliothekskonstruktion über PCR-basierte, PCR-freie und Low-Input-Protokolle durch. Die Methode wird basierend auf der Qualität, der Menge und dem Projekttyp der Probe ausgewählt.
Spezielle ProbenexpertiseWir haben Protokolle zur Bibliothekskonstruktion für FFPE, cfDNA, Einzelzellen und Proben mit extrem niedrigem Input validiert, mit optimierten Adapterverhältnissen, SPRI-Auswahlbereichen und PCR-Zyklusanzahlen für jeden Input-Typ.
QC-StandardsJede Bibliothek unterliegt einer qPCR-Quantifizierung, einer Bioanalyzer-Spurenanalyse und einer Bestätigung der Größenverteilung. Bibliotheken, die die in diesem Leitfaden beschriebenen Akzeptanzkriterien nicht erfüllen, werden markiert und erneut vorbereitet, bevor sie zur Sequenzierung übergehen.
Für weitere Details erkunden Sie unser NGS-Dienstleistungen oder kontaktieren Sie unser Team für projektspezifische Empfehlungen.
Häufig gestellte Fragen
Was ist die optimale Fragmentierungsmethode für den Bau von WGS-Bibliotheken?
Mechanische Fragmentierung (Covaris) erzeugt die gleichmäßigste Abdeckung über den GC-Gehalt mit der geringsten Verzerrung. Enzymatische Fragmentierung liegt dicht dahinter und bietet eine höhere Durchsatzrate. Tagmentation wird für WGS aufgrund der GC-Verzerrung in Regionen mit niedrigem GC-Gehalt nicht empfohlen.
Welches Verhältnis von Adapter zu Insert sollte ich für den Bau einer Standardbibliothek verwenden?
Für Eingaben von 100–500 ng wird ein Adapter-zu-Insert-Molverhältnis von 20:1 bis 30:1 empfohlen. Für höhere Eingaben (>500 ng) sind 10:1 bis 20:1 geeignet; für niedrigere Eingaben kann bis zu 50:1 erforderlich sein.
Wie wähle ich das richtige Verhältnis von SPRI-Perlen für meine Zielfragmentgröße aus?
Für Fragmente von 300–500 bp (Standard-WGS) verwenden Sie eine doppelseitige Auswahl mit einem ersten Perlenverhältnis von 0,6× und einem zweiten Verhältnis von 0,7×. Für Fragmente von 200–300 bp (Amplicon, cfDNA) verwenden Sie 0,6×/0,8×. Für größere Fragmente (500–800 bp) verwenden Sie 0,5×/0,6×.
Wie viele PCR-Zyklen sollte ich für meine Bibliothekskonstruktion verwenden?
Für 100 ng–1 µg Eingabe: 4–8 Zyklen. Für 10–100 ng: 8–10 Zyklen. Für <10 ng: 10–14 Zyklen. Verwenden Sie die minimale Anzahl, die einen ausreichenden Ertrag liefert.
Was bestimmt, ob eine Bibliothek die Qualitätskontrolle besteht?
Schlüsselpasskriterien: qPCR-Konzentration >2 nM, Adapter-Dimer-Gehalt <5% der gesamten Bibliotheksmasse, durchschnittliche Fragmentgröße innerhalb von ±10% des Ziels und qPCR-zu-Qubit-Verhältnis zwischen 0,5 und 2,0.
Wie unterscheide ich Adapter-Dimere von kurzen Bibliotheksfragmenten in einem Bioanalyzer-Trace?
Adapter-Dimeren erscheinen als ein scharfer Peak bei etwa 120–140 bp. Kurze Bibliotheksfragmente erzeugen einen breiteren Peak in demselben Bereich, jedoch mit einer anderen Form. Wenn der fragliche Peak <5% der Gesamtmasse ausmacht, ist es unwahrscheinlich, dass er die Sequenzierleistung erheblich beeinträchtigt.
Warum ist mein Bibliotheksausstoß niedriger als erwartet?
Die häufigsten Ursachen sind: überschätzte Eingangs-DNA-Konzentration (verwenden Sie einen fluorometrischen Test, nicht die UV-Spektrophotometrie), unzureichendes Verhältnis von Adaptern zu Insert, oder übermäßige Bead-Reinigung, die zu viel Material entfernt hat.
Kann ich dasselbe Bibliothekskonstruktionsprotokoll für RNA-Seq und WGS verwenden?
Nein. Die RNA-seq-Bibliothekskonstruktion erfordert die Umkehrtranskription, cDNA-Fragmente und die spezifische Einfügung von Adaptern. Die WGS-Bibliothekskonstruktion verwendet die Fragmentierung von DNA, gefolgt von der Ligation von Adaptern. Es handelt sich um grundlegend unterschiedliche Arbeitsabläufe.
Was ist die minimale DNA-Eingabe für den Standardbibliotheksbau?
Standard-PCR-basierte Kits können mit Eingaben von bis zu 0,1 ng arbeiten, aber Ertrag und Komplexität werden unter 1 ng unzuverlässig. Für Eingaben unter 10 ng wird ein speziell für die Konstruktion von Low-Input-Bibliotheken entwickeltes Kit empfohlen.
Wie beeinflusst die Bisulfit-Konversion den Bibliotheksaufbau für die Ganzgenom-Bisulfit-Sequenzierung?
Bisulfitbehandlung wandelt unmethylierte Cytosine in Uracil um, das während der Sequenzierung als Thymin gelesen wird. Dies reduziert die Sequenzkomplexität und erfordert spezialisierte Bibliothekskonstruktionsprotokolle mit methylisierten Adaptern und bisulfitkompatiblen Polymerasen.
Was ist der häufigste Grund für eine niedrige Clusterdichte aus einer gut quantifizierten Bibliothek?
Die häufigste versteckte Ursache ist eine schlechte Effizienz der Adapterligatur. Die Bibliothek besteht den Qubit-Quantifizierungs-Test (der gesamtes dsDNA misst), aber viele Moleküle haben an beiden Enden keine funktionalen Adapter. Das gleichzeitige Durchführen von qPCR- und Qubit-Messungen und der Vergleich des Verhältnisses ist die beste Diagnosemethode.
Wie wähle ich zwischen einseitiger und zweiseitiger SPRI-Reinigung für meine Bibliothek?
Einseitige Reinigung ist schneller und ausreichend für Anwendungen, bei denen das Entfernen von Adapter-Dimeren das Hauptziel ist und große Fragmente weniger von Bedeutung sind. Eine zweiseitige Reinigung wird für WGS und größenkritische Anwendungen empfohlen, bei denen sowohl große als auch kleine Fragmente kontrolliert werden müssen.
Kann ich die gleichen Bibliothekskonstruktionsparameter für FFPE-DNA wie für DNA aus frisch gefrorenem Gewebe verwenden?
Nein. FFPE-DNA ist typischerweise degradiert (durchschnittliche Fragmentgröße 200–400 bp) und enthält beschädigte Basen. Der Aufbau der Bibliothek für FFPE sollte einen Vorreparaturschritt mit Uracil-DNA-Glykosylase, weniger PCR-Zyklen (um die Amplifikation von Artefakten zu vermeiden) und ein breiteres Größenwahlfenster verwenden, das die kürzere, heterogenere Fragmentverteilung berücksichtigt.
Was sind die optimalen Lagerbedingungen für fertiggestellte Bibliotheken?
Fertige Bibliotheken sollten bei -20°C in niedrig-adsorbierenden Röhrchen gelagert werden. Unter diesen Bedingungen sind die Bibliotheken mindestens 6 Monate stabil. Vermeiden Sie wiederholte Gefrier-Tau-Zyklen, indem Sie die Bibliotheken in Einmalvolumina aliquotieren.
Wie bestimme ich, ob mein Bibliothekskonstruktionsprotokoll optimiert werden muss?
Der zuverlässigste Indikator ist das Verhältnis von qPCR zu Qubit. Ein Verhältnis, das konstant über 3,0 liegt, weist darauf hin, dass ein erheblicher Teil der quantifizierten DNA kein funktionales Bibliotheksmaterial ist. Weitere Anzeichen sind: konstant niedrige Cluster-Dichte trotz angemessener Lade-Konzentration, Duplikationsraten über 30 % oder Adapter-Dimer-Spitzen über 10 % in Bioanalyzer-Spuren.
Nur für Forschungszwecke.
Referenzen:
- Optimierung von DNA-Fragmentierungstechniken. Diagnostik2025;15:2294.
- Optimierung der enzymatischen Fragmentierung. BMC Genomik. 2022;23:89.
- Technische Verbesserung der NGS in klinischen Anwendungen – Optimierung der Adapterligatur. Praktische Laboratoriumsmedizin2025;40:e00430.
- Eine vergleichende Analyse von Bibliotheksvorbereitungsansätzen für Proben mit geringem Input. BMC Genomics2018;19:763.
- Multiplex-PCR mit niedriger Zyklusanzahl zur Bibliothekskonstruktion. Elektrophorese2024;45:1255-1264.


