
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Umfassender Überblick über m6A-Detektionsmethoden
Da unser Verständnis von RNA-Epigenetik Fortschritte, die Bedeutung einer genauen Unterscheidung N6-Methyladenosin (m6A) Modifikationen Innerhalb von RNA wird zunehmend bedeutend. m6A, die vorherrschende interne Modifikation, die in eukaryotischer messenger RNA (mRNA) vorkommt, spielt eine entscheidende Rolle in verschiedenen biologischen Prozessen, die von der Stabilität der mRNA über Spleißen, Translation bis hin zur zellulären Differenzierung reichen. Daher wird die Notwendigkeit, robuste und präzise Methoden zur m6A-Detektion zu entwickeln, als entscheidend angesehen, um unser Verständnis der RNA-Epigenetik und ihrer Auswirkungen auf Gesundheit und Krankheit voranzutreiben. In diesem umfassenden Überblick begeben wir uns auf eine Erkundung verschiedener m6A-Detektionsmodalitäten und erläutern deren zugrunde liegende Prinzipien, Vorteile und Einschränkungen, wobei wir besonderes Augenmerk auf die jüngsten Fortschritte in diesem Bereich legen.
Sie könnten interessiert sein an
Antikörperbasierte Methoden
Frühe Techniken wie die m6A-Immunpräzipitation (m6A-IP) basieren auf Antikörpern, um RNA-m6A-Modifikationen zu profilieren. Solche Methoden nutzen die Selektivität der Antikörper, um m6A-modifizierte RNA aus Gesamtnachweisen anzureichern. Dennoch haben diese Methoden Einschränkungen. Die Spezifität der Antikörper und unspezifische Bindungen können zu falsch positiven Ergebnissen führen, was die Nachweisgenauigkeit beeinträchtigt. Darüber hinaus kann die Abhängigkeit von Antikörpern zu Batch-Variabilität führen, was die Reproduzierbarkeit potenziell beeinträchtigt.
Chemische Methoden
Chemiebasierte Methoden bieten praktikable Alternativen zur m6A-Detektion und umgehen die Abhängigkeit von Antikörpern. Besonders hervorzuheben ist die m6A-selektive chemische Markierung (m6A-SEAL), die spezifische chemische Sonden verwendet, um m6A-Stellen innerhalb von RNA zu kennzeichnen. Die anschließende Sequenzierung ermöglicht eine präzise Identifizierung von m6A-Modifikationen auf Nukleotid-Ebene. Ähnlich kombiniert miCLIP (m6A Einzel-Nukleotid-Auflösung UV-Crosslinking und Immunpräzipitation) UV-Crosslinking, Immunpräzipitation und chemische Modifikation und ermöglicht eine hochauflösende Kartierung von m6A-Stellen. Chemische Ansätze bieten Vorteile wie eine verringerte Abhängigkeit von Antikörpern und eine verbesserte Auflösung. Dennoch könnte ihre Anwendung durch komplexe experimentelle Protokolle und Anfälligkeit für Hintergrundrauschen eingeschränkt sein, was die weitverbreitete Anwendung limitiert.
Enzymbasierte Methoden
Enzymbasierte Methoden erweisen sich als vielversprechende Ansätze für m6A-Detektionder Nutzung von ingenieurtechnisch entwickelter Enzymkatalyse zur Modifikation von m6A-beladenen RNA-Molekülen. Ein Beispiel in diesem Bereich ist DART-seq (Deaminierung von Adenosinen, RNA-Zielgerichtete Sequenzierung), das Fusionsproteine verwendet, die maßgeschneiderte Domänen für die gezielte RNA-Modifikation enthalten. Das DART-Fusionsprotein, das eine RNA-bindende Domäne mit einer Editierungsdomäne integriert, ermöglicht die präzise Identifizierung von m6A-Stellen mit einer Auflösung auf Einzel-Nukleotid-Ebene. Jüngste Fortschritte in DART-seq, einschließlich verbesserter Fusionsproteine mit erhöhter m6A-Erkennung, steigern die Effizienz der m6A-Kartierung. Enzymgesteuerte Ansätze bieten eine erhöhte Spezifität und Sensitivität im Vergleich zu antikörperbasierten Modalitäten, was sie unverzichtbar macht in RNA-Epigenetik Untersuchungen.
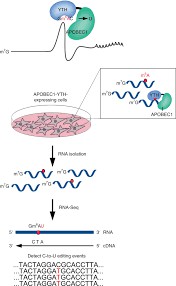 DART-seq
DART-seq
Sequenzierungsbasierte Methoden
Hochdurchsatz-Sequenzierung Methoden, veranschaulicht durch MeRIP-seq (Methylierte RNA-Immunpräzipitation-Sequenzierung) und Nanopore-RNA-Sequenzierungsind entscheidend für die Aufklärung von m6A-Modifikationen über die TranskriptomDurch die Nutzung dieser Ansätze gewinnen Forscher tiefgreifende Einblicke in die Verteilung und Dynamik von m6A-Modifikationen innerhalb von RNA-Molekülen.
MeRIP-seq: Entschlüsselung der m6A-Verteilung mit Antikörper-Immunpräzipitation
MeRIP-seq nutzt die Affinität von Antikörpern für m6A, um m6A-modifizierte RNA-Fragmente selektiv aus Gesamt-RNA-Proben anzureichern. Diese Methode bietet Forschern ein Mittel, um m6A-Stellen über die Transkriptom mit erheblicher Auflösung. Durch die Fällung von m6A-modifizierten RNA-Fragmenten bietet MeRIP-seq einen umfassenden Überblick über die Verteilung von m6A, was die Erkennung von m6A-modifizierten Regionen und deren funktionaler Relevanz erleichtert. Allerdings kann MeRIP-seq auf Herausforderungen wie Antikörperspezifität und unspezifische Bindung stoßen, was die Genauigkeit der m6A-Erkennung beeinträchtigen könnte.
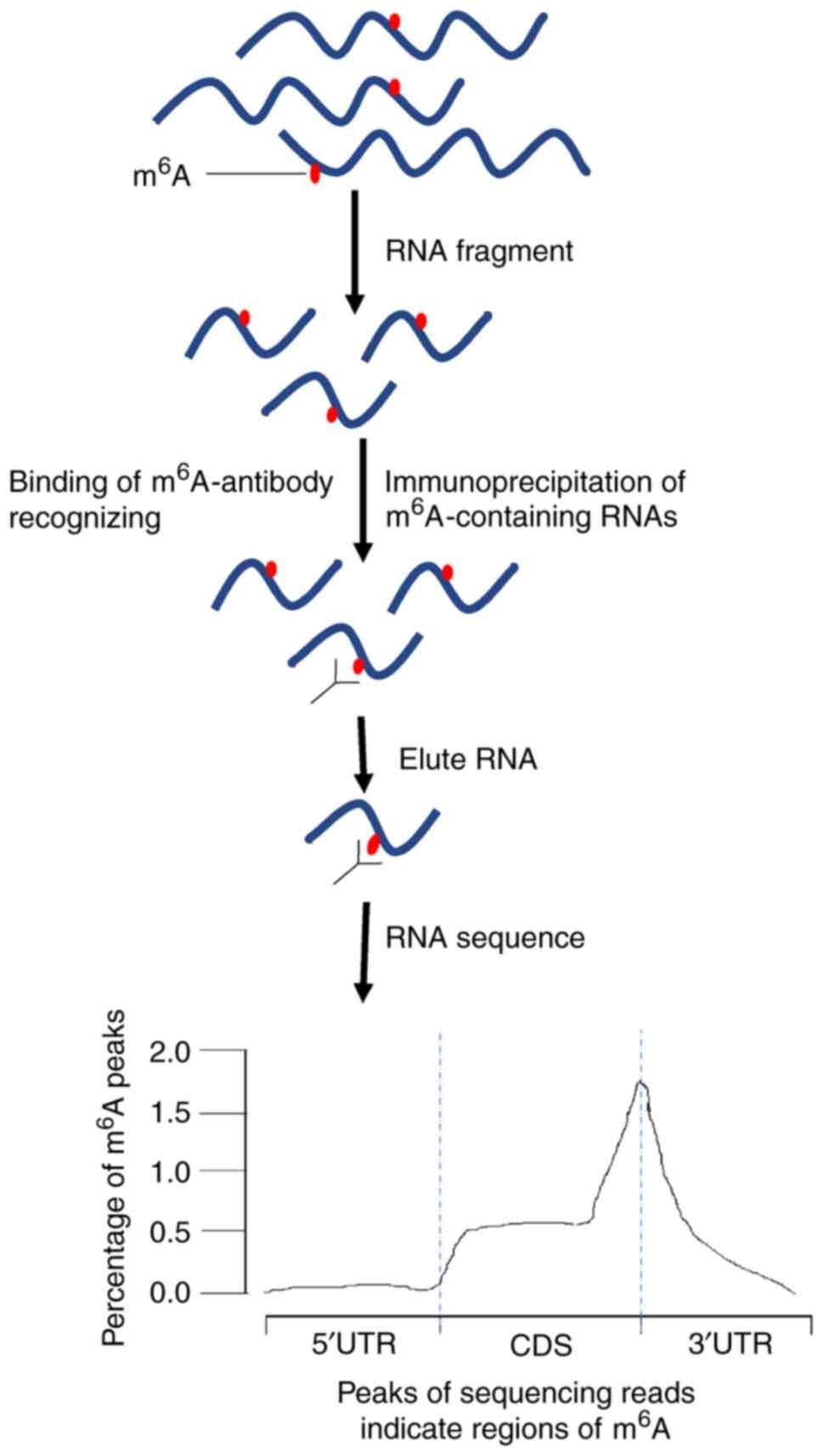 MeRIP-seq
MeRIP-seq
Nanopore-RNA-Sequenzierung: Direkte Sequenzierung von RNA-Molekülen zur m6A-Detektion
Nanopore-RNA-Sequenzierung stellt einen Paradigmenwechsel in der m6A-Detektion dar und ermöglicht die direkte Analyse von RNA-Molekülen auf m6A-Modifikationen ohne vorherige Anreicherung oder Markierung. Diese Technologie nutzt Nanoporen in einer Membran, um Veränderungen des elektrischen Stroms zu erkennen, während RNA hindurchtritt. Die charakteristischen elektrischen Signaturen, die mit m6A-Modifikationen verbunden sind, ermöglichen die präzise Identifizierung von m6A-modifizierten Nukleotiden innerhalb von RNA-Sequenzen. Die Nanoporen-RNA-Sequenzierung bietet Vorteile wie vereinfachte experimentelle Verfahren und die Echtzeit-Erfassung dynamischer m6A-Modifikationen. Dennoch stellt die Bewältigung von Herausforderungen wie Signalrauschen und Datenanalyse weiterhin ein aktives Forschungs- und Entwicklungsfeld dar.
Für weitere Informationen siehe "Vergleich der M6A-Sequenzierungsmethoden"
Massenspektrometriebasierte Methoden
Massenspektrometrie-basierte Methoden, exemplifiziert durch m6A-LAIC-seq (m6A-Level und Isoform-Charakterisierung-Sequenzierung), stellen einen ergänzenden Ansatz zur m6A-Detektion dar. Diese Techniken beinhalten die direkte Analyse von RNA-Molekülen auf das Vorhandensein von m6A unter Verwendung von Massenspektrometrie. Bei m6A-LAIC-seq werden RNA-Moleküle zunächst enzymatisch in Nukleoside zerlegt, gefolgt von einer Flüssigkeitschromatographie, gekoppelt mit Massenspektrometrie (LC-MS), zur Quantifizierung des m6A-Levels. Während massenspektrometriebasierte Ansätze quantitative Daten über die Häufigkeit von m6A liefern, sind sie in der Lage, die genauen Positionen der m6A-Modifikationen innerhalb der RNA-Sequenzen zu bestimmen.
 m6A-LAIC-seq
m6A-LAIC-seq
Hybride Methoden
Hybride Methoden vereinen verschiedene Detektionsstrategien, um die Einschränkungen einzelner Ansätze zu überwinden und eine umfassende m6A-Kartierung zu fördern. Zum Beispiel kombiniert m6A-REF-seq (m6A-RECombination und Fragmentierungssequenzierung) antikörperbasierte Immunpräzipitation mit enzymatischer Fragmentierung und Sequenzierung, um m6A-Stellen mit Einzel-Nukleotid-Präzision zu identifizieren. Ebenso kombiniert m6A-REF-seq2 chemische Markierung mit Immunpräzipitation und Sequenzierung, um die Genauigkeit der m6A-Detektion zu verbessern. Hybride Methoden bieten Vorteile wie erhöhte Sensitivität, Spezifität und Auflösung im Vergleich zu Einzelmethoden, allerdings möglicherweise auf Kosten einer erhöhten experimentellen Komplexität und der Komplexität der Datenanalyse.
Bioinformatische Methoden
Bioinformatische Methoden eine entscheidende Rolle bei der Analyse von Daten aus m6A-Detektionsexperimenten und der Identifizierung von m6A-modifizierten Stellen innerhalb von RNA-Sequenzen. Diese Ansätze beinhalten die Entwicklung von computergestützten Algorithmen und Softwaretools zur Verarbeitung von Rohsequenzierungsdaten, zur Ausrichtung von Reads an Referenzgenomen oder Transkriptomikund skizzieren m6A-Spitzen basierend auf Anreicherungsmustern. Prominente bioinformatische Werkzeuge für die m6A-Analyse umfassen HOMER, MACS und MeTDiff. Durch die Nutzung verschiedener statistischer Modelle und maschineller Lernalgorithmen identifizieren diese Werkzeuge geschickt m6A-modifizierte Regionen und sagen potenzielle m6A-Stellen innerhalb von RNA-Sequenzen mit Präzision voraus. Bioinformatische Methoden sind unverzichtbare Hilfsmittel zur Entschlüsselung experimenteller Ergebnisse und zur Aufdeckung der funktionalen Implikationen von m6A-Modifikationen in der Genregulation und zellulären Prozessen.
Vergleich der m6A-Detektionsmethoden
| Methode | Prinzip | Merkmale | Vorteile | Nachteile |
| Antikörperbasiert | Basiert auf Antikörpern, um m6A-modifizierte RNA-Fragmente aus Gesamt-RNA-Proben anzureichern. | - Verwendet spezifische Antikörper, die auf m6A-Modifikationen abzielen - Immunpräzipitation-basierte Anreicherung von m6A-modifizierten RNA-Fragmenten aus Gesamt-RNA-Proben | - Schnelle Identifizierung von m6A-Modifikationen auf Transkriptomebene - Kann quantitative Informationen über die m6A-Häufigkeit bereitstellen | - Die Auswahl der Antikörper ist entscheidend und kann zu falsch positiven oder falsch negativen Ergebnissen führen. - Die Batch-zu-Batch-Variabilität in der Antikörperqualität kann die Reproduzierbarkeit beeinträchtigen. |
| Chemiebasiert | Verwendet spezifische chemische Sonden, um m6A-Stellen in RNA zu markieren. | - Chemische Kennzeichnung von m6A-Stellen in RNA-Molekülen mithilfe chemischer Sonden - Ermöglicht hochauflösende Kartierung von m6A-Modifikationen | - Unabhängig von Antikörpern, Verringerung der Variabilität in der Reproduzierbarkeit - Bietet eine höhere Auflösung und Empfindlichkeit im Vergleich zu antikörperbasierten Methoden. | - Komplexe experimentelle Verfahren können spezialisiertes Fachwissen erfordern - Hintergrundgeräusche von chemischen Reaktionen können die Datengenauigkeit beeinträchtigen |
| Enzymatisch basierend | Nutzt die katalytische Aktivität von konstruierten Enzymen, um m6A-modifizierte RNA-Moleküle zu modifizieren. | - Fusionsproteine mit konstruierten Domänen zur gezielten RNA-Modifikation - Ermöglicht die Abbildung von m6A mit Einzel-Nukleotid-Auflösung | - Erhöhte Spezifität und Sensitivität im Vergleich zu antikörperbasierten Methoden - Bietet eine Einzel-Nukleotid-Auflösung für präzises m6A-Mapping | - Die Entwicklung und Optimierung von konstruierten Enzymen kann herausfordernd sein - Erfordert Validierung und Optimierung für jede spezifische Anwendung |
| Sequenzierungsbasiert | Nutzen von Hochdurchsatz-Sequenzierungstechnologien zur Profilierung von m6A-Modifikationen. | - Vertraut auf Hochdurchsatz-Sequenzierungsplattformen, um m6A-Modifikationen im gesamten Transkriptom zu profilieren - Bietet genomweite m6A-Karten und Einblicke in die Verteilung und Dynamik von m6A | - Ermöglicht die Identifizierung von m6A-Modifikationen im großen Maßstab - Bietet Einblicke in die Verteilung und Dynamik von m6A | - Unterschiedliche Auflösung und Spezifität bei verschiedenen sequenzierungsbasierten Methoden - Experimentelle Komplexität kann variieren, was sorgfältige Überlegung und Optimierung erfordert. |
| Massenspektrometriebasiert | Analysiert RNA-Moleküle direkt auf das Vorhandensein von m6A-Modifikationen mittels Massenspektrometrie. | - Direkte Analyse von RNA-Molekülen auf das Vorhandensein von m6A-Modifikationen mittels Massenspektrometrie - Bietet quantitative Informationen über die m6A-Häufigkeit | - Quantitative Bewertung der m6A-Häufigkeit und relativen Werte - Ergänzend zu sequenzierungsbasierten Methoden zur Validierung und Bestätigung | - Eingeschränkte Fähigkeit, spezifische m6A-Modifikationsstellen innerhalb von RNA-Sequenzen genau zu bestimmen - Erfordert spezielle Ausrüstung und Fachkenntnisse für die Massenspektrometrie-Analyse |
| Hybrid | Integriert verschiedene Erkennungsstrategien, um Einschränkungen zu überwinden. | - Kombination mehrerer Methoden zur Verbesserung von Sensitivität, Spezifität und Auflösung - Bietet komplementäre Stärken der einzelnen Methoden | - Verbesserte Sensitivität, Spezifität und Auflösung im Vergleich zu Einzelmethoden - Überwindet die Einschränkungen einzelner Methoden | - Komplexe experimentelle Verfahren können zusätzliche Optimierung und Validierung erfordern - Die Datenintegration und -analyse kann aufgrund der hybriden Natur des Ansatzes herausfordernd sein |
| Bioinformatik | Analysiert Daten aus m6A-Detektionsexperimenten mithilfe von computergestützten Algorithmen. | - Nutzt rechnergestützte Algorithmen und Werkzeuge zur Verarbeitung und Analyse von Rohsequenzdaten - Identifiziert m6A-modifizierte Stellen innerhalb von RNA-Sequenzen basierend auf Anreicherungsmustern | - Wesentlich für die Interpretation experimenteller Ergebnisse und das Verständnis der funktionalen Rollen von m6A-Modifikationen - Erleichtert die Dateninterpretation und -integration | - Die Abhängigkeit von computergestützten Methoden kann Verzerrungen und Fehler einführen - Erfordert Fachkenntnisse in der Bioinformatik für eine genaue Datenanalyse und -interpretation |
Diese detaillierte Tabelle bietet einen umfassenden Vergleich verschiedener m6A-Detektionsmethoden und hebt deren zugrunde liegende Prinzipien, Eigenschaften, Vorteile und Nachteile mit einem spezialisierten Fokus hervor.
Zusammenfassung
Zusammenfassend lässt sich sagen, dass das Reich der m6A-Detektion Die Methoden sind vielschichtig, wobei jeder Ansatz unterschiedliche Vorteile und Herausforderungen mit sich bringt. Antikörperbasierte Methoden ermöglichen eine schnelle Identifizierung von m6A im gesamten Transkriptom, können jedoch auf Bedenken hinsichtlich der Antikörperspezifität stoßen. Chemische Techniken bieten eine höhere Auflösung und Empfindlichkeit, erfordern jedoch eine sorgfältige Optimierung und können mit Hintergrundgeräuschen konfrontiert sein. Enzymatische Methoden bieten eine erhöhte Spezifität und Sensitivität, erfordern jedoch eine strenge Validierung für jede Anwendung. Sequenzierungsbasierte Modalitäten liefern genomweite m6A-Karten, variieren jedoch in Auflösung und experimenteller Komplexität. Massenspektrometriebasierte Ansätze ermöglichen eine quantitative Bewertung der m6A-Häufigkeit, scheitern jedoch daran, spezifische Modifikationsstellen genau zu bestimmen. Hybride Strategien kombinieren verschiedene Taktiken, um Einschränkungen zu überwinden, erfordern jedoch zusätzliche Optimierung. Bioinformatische Methoden sind für die Datenanalyse und -interpretation unerlässlich und erfordern Kenntnisse in der computationalen Biologie. Forscher müssen diese Aspekte sorgfältig bewerten, um die optimale Methode für ihre Forschungsziele auszuwählen. Während die m6A-Detektion voranschreitet, bietet CD Genomics kontinuierlich umfassende Lösungen, um die RNA-Epigenetikforschung voranzutreiben.
Referenzen:
- Capitanchik, C., Toolan-Kerr, P., Luscombe, N. M., & Ule, J. Wie identifizieren Sie m6A-Methylierung in Transkriptomen mit hoher Auflösung? Ein Vergleich aktueller Datensätze. Grenzen der Genetik, (2020). 11, 398.
- Fan, C., Ma, Y., Chen, S., Zhou, Q., Jiang, H., & Zhang, J. Umfassende Analyse der transkriptomweiten m6A-Methylierungsmodifikation bei Leberfibrose-Mäusen durch Hochdurchsatz-m6A-Sequenzierung. Frontiers in Zell- und Entwicklungsbiologie, (2021). 9, 767051.
- Zhu, H., Yin, X., Holley, C. L., & Meyer, K. D. Verbesserte Methoden zur Deaminierungsbasierten m6A-Detektion. Frontiers in Zell- und Entwicklungsbiologie, (2022). 10, 888279.
- Liu, H., Begik, O., Lucas, M.C. et al. Genauige Erkennung von m6A RNA-Modifikationen in nativen RNA-Sequenzen. Nat Commun 10, 4079 (2019).
- Hendra, C., Pratanwanich, P.N., Wan, Y.K. et al. Nachweis von m6A aus direkter RNA-Sequenzierung unter Verwendung eines Multiple-Instance-Learning-Frameworks. Nat Methoden 19, 1590–1598 (2022).
- Meyer, K.D. DART-seq: eine antikörperfreie Methode zur globalen m6A-Detektion. Nat Methoden 16, 1275–1280 (2019)
- Zhu W, Wang JZ, Xu Z, Cao M, Hu Q, Pan C, Guo M, Wei JF, Yang H. Nachweis von N6-Methyladenosin-Modifikationsrückständen (Überblick). Int J Mol Med. 2019


