
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Was ist Next Generation Sequencing (NGS)?
Die Next-Generation-Sequenzierung (NGS) ist keine einzelne Technologie. Es handelt sich um ein Projektsystem, das Probenvorbereitung, Bibliothekskonstruktion, Plattformauswahl, Sequenzierungsparameter und bioinformatische Analyse integriert. Die Wahl jedes einzelnen Elements bestimmt, ob die endgültigen Daten die ursprüngliche biologische Frage beantworten können.
Dieser Leitfaden richtet sich an Forscher, die bereits verstehen, was NGS ist, und einen entscheidungsorientierten Rahmen für die Planung ihres nächsten Sequenzierungsprojekts benötigen. Er behandelt, wie man die richtige Sequenzierungsstrategie basierend auf Forschungszielen, Probenmerkmalen, Plattformfähigkeiten und Datenqualitätsanforderungen auswählt. Der Fokus liegt auf umsetzbarer Projektgestaltungslogik und nicht darauf, grundlegende NGS-Definitionen zu wiederholen, die bereits in bestehenden Ressourcen gut abgedeckt sind.
Von Sequenzierungstechnologie zu Projektentwurfssystem
NGS unterscheidet sich grundlegend von der Sanger-Sequenzierung in drei Aspekten, die jede Projektgestaltungsentscheidung beeinflussen. Erstens liest NGS Millionen bis Milliarden von DNA-Fragmenten parallel und erzeugt Datenmengen, die eine rechnergestützte Infrastruktur für die Verarbeitung und Interpretation erfordern. Zweitens erzeugt NGS kurze oder lange Reads, die an ein Referenzgenom ausgerichtet oder de novo assembliert werden müssen – der Typ des Reads bestimmt, welche biologischen Fragen behandelt werden können. Drittens hängt die Datenqualität von NGS von der Sequenzierungstiefe, der Gleichmäßigkeit der Abdeckung, den Fehlermodellen und der Qualität der Referenzdatenbank ab, nicht nur von der Genauigkeit der Plattform.
Forscher, die ein NGS-Projekt bewerten, sollten sich vor der Auswahl einer Plattform oder Dienstleister auf fünf praktische Fragen konzentrieren:
- Kann mein Probenart und -qualität die beabsichtigte Analyse unterstützen?
- Sollte ich eine Short-Read-, Long-Read- oder Hybrid-Strategie wählen?
- Wie viel Sequenzierungstiefe benötige ich für zuverlässige Ergebnisse?
- Welche QC-Metriken sollte ich in jeder Phase verfolgen?
- Welche biologischen Fragen kann die resultierende Daten beantworten, und welche Fragen bleiben außerhalb ihres Umfangs?
Diese Fragen bilden das Rückgrat des Projektentwurfsrahmens, der in diesem Leitfaden beschrieben wird. Für Forscher, die ihr erstes NGS-Projekt beginnen oder eine neue Anwendung bewerten, umfassende NGS-Dienstleistungen Bieten Sie fachkundige Anleitung zur Versuchsplanung und Auswahl von Strategien.
 Abbildung 1. NGS-Projektdesign-Stack — von der Probe zur biologischen Interpretation
Abbildung 1. NGS-Projektdesign-Stack — von der Probe zur biologischen Interpretation
Fünfschichtiger NGS-Projektdesign-Stack, der den Fortschritt von der biologischen Fragestellung über die Probenvorbereitung, den Bibliotheksaufbau, die Plattformwahl, die Sequenzierungsparameter und die bioinformatische Analyse bis zur biologischen Interpretation zeigt.
Von der Forschungsfrage zur NGS-Strategie — Ein Entscheidungsrahmen
Der häufigste Fehler bei der Planung von NGS-Projekten besteht darin, mit der Plattform zu beginnen, anstatt mit der biologischen Fragestellung. Die richtige Sequenzierungsstrategie hängt davon ab, was die Forschung zu entdecken, zu messen oder zu vergleichen beabsichtigt.
Klassifizierung von Forschungszielen nach NGS-Anforderungen: Der erste Schritt bei der Planung eines NGS-Projekts besteht darin, das Forschungsziel nach der Art der benötigten biologischen Informationen zu klassifizieren. Jede Zielart hat spezifische Anforderungen an die Sequenzierungsparameter, die die Wahl der Plattform, der Tiefe und der Analyse-Pipeline bestimmen.
| Forschungsziel | Bessere NGS-Strategie | Schlüsselgestaltungsvariable | Risiko bei Fehlanpassung |
|---|---|---|---|
| SNP / kleine InDel-Erkennung | Kurzzeit-WGS / WES / gezielte Panel | Tiefe, Mapping-Qualität, Duplikationsrate | Anrufe mit geringer Zuversicht |
| Entdeckung struktureller Varianten | Langzeit- oder Hybrid-WGS | Lese-Länge, Molekülintegrität | Verpasste SVs |
| De-novo-Genomassemblierung | PacBio HiFi / ONT / Hybrid | N50, Abdeckung, Heterozygotie | Fragmentierte Montage |
| Differenzielle Expression | RNA-Seq | RNA-Integrität, Bibliothekstyp, biologische Replikate | Falsche biologische Interpretation |
| Mikrobiom-Profiling | Amplicon- oder Shotgun-Metagenomik | Markerregion, Datenbank, Hosterschöpfung | Voreingenommener taxonomischer Profil |
Entscheidungslogik: Wenn ein hochwertiges Referenzgenom für die Zielart verfügbar ist, ist die Kurzlesesequenzierung der kosteneffektivste Ansatz für die meisten Anwendungen zur Variantenentdeckung und -quantifizierung. Der Nachteil ist eine begrenzte Auflösung in repetitiven Regionen. Wenn die Forschung erfordert, dass repetitive Regionen länger als die Lese-Länge aufgelöst werden, große strukturelle Varianten (Löschungen, Einfügungen, Inversionen über 50 bp) erkannt werden oder ein Genom de novo assemblieren muss, ist die Langlesesequenzierung notwendig, trotz ihrer höheren Kosten pro Basenpaar und strengeren Anforderungen an die Proben.
Für Projekte, die sowohl Genauigkeit als auch Kontinuität erfordern, bieten hybride Strategien, die Kurz- und Langleseplattformen kombinieren, das beste Gleichgewicht. Eine typische hybride Genomassemblierung verwendet PacBio HiFi- oder Nanopore-Lesungen für den Contig-Bau und Illumina-Kurzlesungen für die Nachbearbeitung und Fehlerkorrektur. Die Kosten dieses Dual-Plattform-Ansatzes sind höher, aber die resultierende Qualitätsverbesserung der Assemblierung rechtfertigt die Investition für Projekte mit hoher Priorität.
Häufige Fehler, die man vermeiden sollte:
- "Wie viele Gigabasen?" ohne zuvor die biologische Frage zu definieren.
- Die Preise von Plattformen zu vergleichen, ohne zu bewerten, ob die resultierenden Daten die beabsichtigte Schlussfolgerung unterstützen können.
- WGS, WES, RNA-seq und Amplicon-Sequenzierung als austauschbare Projekttypen behandeln
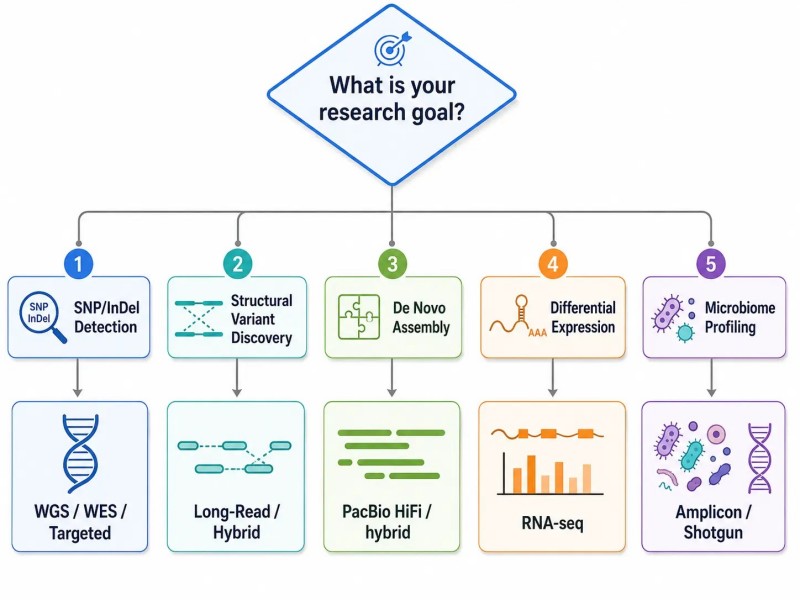 Abbildung 2. Forschungsfrage zum Entscheidungsbaum der NGS-Strategie
Abbildung 2. Forschungsfrage zum Entscheidungsbaum der NGS-Strategie
Bildunterschrift: Entscheidungsbaum, der fünf häufige Forschungsziele – SNP-Erkennung, Entdeckung struktureller Varianten, de novo Assemblierung, differentielle Expression und Mikrobiom-Profiling – ihren optimalen NGS-Strategien mit wichtigen Entwurfsvariablen und Risiken der Fehlanpassung zuordnet.
Plattformauswahl — Wie man die Eignung einer Plattform für Ihr Projekt bewertet
Die Auswahl der Plattform betrifft nicht die Rangordnung von Technologien, sondern das Abgleichen ihrer Eigenschaften mit den spezifischen Anforderungen eines Forschungsprojekts. Die ABRF-Studie zur Next-Generation-Sequenzierung hat gezeigt, dass verschiedene Plattformen messbar unterschiedliche Ergebnisse in Bezug auf die Konsistenz der Abdeckung, Fehlerquoten und die Leistung bei der Variantenerkennung liefern. Diese Unterschiede bedeuten, dass die Wahl der Plattform direkt beeinflusst, welche biologischen Erkenntnisse entdeckt werden können.
Wichtige projektbezogene Überlegungen: Für Projekte, bei denen Durchsatz und Genauigkeit pro Base die Hauptanforderungen sind – SNV-Erkennung, RNA-seq-Quantifizierung, Exom-Sequenzierung und gezielte Panels – ist die Kurzlesesequenzierung der am weitesten verbreitete Ansatz. NGS-Dienstleistungen Das Portfolio umfasst mehrere Short-Read-Plattformen, um den Durchsatz an die Projektgröße anzupassen.
Für Projekte, die die Auflösung genomischer Regionen erfordern, die länger sind als die Leselänge—de novo Assemblierung, Erkennung struktureller Varianten und Sequenzierung vollständiger Transkripte—sind Langlese-Plattformen notwendig, trotz höherer Kosten pro Basenpaar und strengerer Anforderungen an die Proben. Eine detaillierte Vergleich von PacBio- und Oxford-Nanopore-Technologien steht Forschern zur Verfügung, die Langzeitoptionen bewerten.
Für Projekte, die sowohl Genauigkeit als auch Kontinuität erfordern – umfassende Genomassemblierung oder vollständige Variantenerkennung – bieten hybride Strategien, die Kurz- und Langzeit-Sequenzierung kombinieren, das beste Gleichgewicht. Dieser Dual-Plattform-Ansatz erfordert eine höhere Gesamtinvestition, liefert jedoch eine Datenqualität, die keine der Plattformen allein erreichen kann.
Häufige Fehler bei der Auswahl von Plattformen: Die Annahme, dass längere Reads universell besser sind, die Annahme, dass kurze Reads nicht zur Analyse von strukturellen Varianten beitragen können, und die Erwartung, dass eine Plattform für alle Projekttypen optimal ist, gehören zu den häufigsten Fehlern bei der Planung von NGS-Projekten.
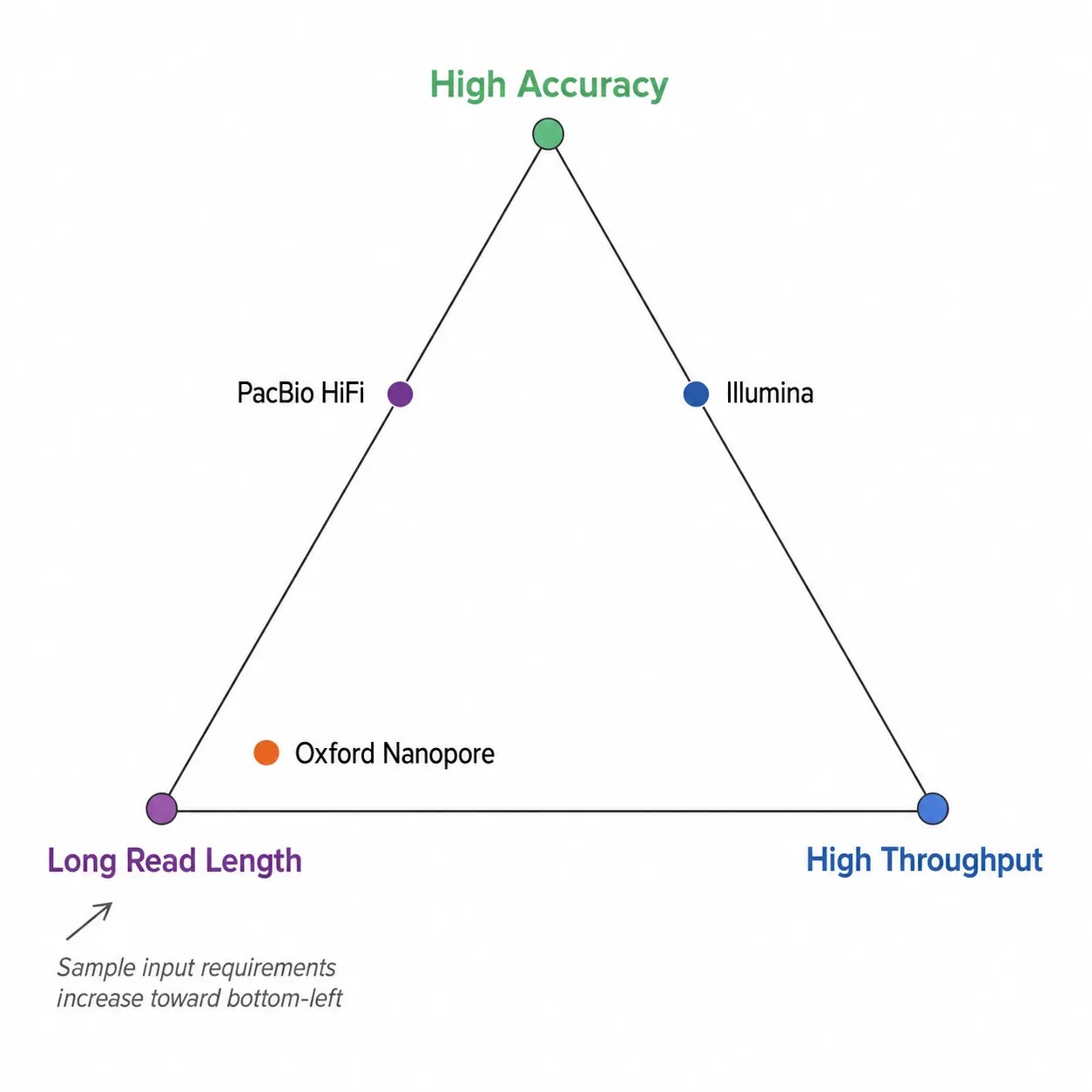 Abbildung 3. Trade-off-Dreieck der NGS-Plattform — Genauigkeit, Lese-Länge, Durchsatz und Probenanforderungen
Abbildung 3. Trade-off-Dreieck der NGS-Plattform — Genauigkeit, Lese-Länge, Durchsatz und Probenanforderungen
Trade-off-Dreieck, das die Wechselbeziehung zwischen Genauigkeit, Lese länge, Durchsatz und Probenanforderungen bei Kurzlese-, Langlese- und hybriden NGS-Plattformen veranschaulicht.
Die Probenqualität ist die erste Einschränkung in jedem NGS-Projekt.
Keine Menge an Sequenzierungstiefe oder ausgeklügelter Bioinformatik kann schlechte Probenqualität ausgleichen. Die Bewertung der Probenqualität sollte der erste Schritt bei der Projektplanung sein, bevor die Plattform oder die Bibliotheksauswahl getroffen wird.
Schlüsselvariablen für DNA-Proben: Eingabemenge (Gesamtmasse und Konzentration), Fragmentgrößenverteilung, Abbaustufe (bewertet durch Gelelektrophorese oder TapeStation), Reinheitsverhältnisse (A260/280, A260/230) und Kontaminanten (Phenol, Ethanol, Salze, Polysaccharide, Häm, Huminsäure) beeinflussen alle die Effizienz des Bibliotheksaufbaus und die Qualität der Sequenzierungsdaten. Eine Probe, die die Konzentrations-QC besteht, aber restliches Phenol enthält, wird im Ligation-Schritt fehlschlagen, da Phenol die DNA-Ligase hemmt. Aus diesem Grund bietet die spektrophotometrische Reinheitsbewertung in Kombination mit fluorometrischer Quantifizierung ein zuverlässigeres Qualitätsbild als jede Methode für sich allein.
Quantitative QC-Grenzwerte für DNA: A260/280 sollte für reines DNA zwischen 1,8 und 2,0 liegen; Werte außerhalb dieses Bereichs deuten auf Protein- oder Phenolkontamination hin. A260/230 sollte zwischen 2,0 und 2,2 liegen; niedrigere Werte deuten auf Rückstände organischer Verbindungen oder Kohlenhydrate hin. Für hochmolekulare DNA, die von Long-Read-Plattformen benötigt wird, sollte die genomische DNA auf einem Gel oder TapeStation-Diagramm ein dominantes Band über 20 kb zeigen, ohne signifikantes Verwischen unter 10 kb.
Schlüsselfaktoren für RNA-Proben: RIN-Score (RIN ≥ 7 für mRNA-seq, RIN ≥ 5 für total RNA-seq), DV200 für FFPE-RNA-Proben, rRNA-Kontaminationsniveau und Gewebeerhaltungsmethode. RNA aus FFPE erfordert spezifische Bibliotheksvorbereitungsprotokolle mit Schritten zur Schadensreparatur, und der erwartete Ertrag ist typischerweise niedriger als bei frisch gefrorenem Gewebe.
Zusätzliche Überlegungen zur Langzeit-Sequenzierung: Die Extraktion von hochmolekularen DNA ist entscheidend. Sanfte Handhabung während der Extraktion, minimale Gefrier-Tau-Zyklen und das Vermeiden von mechanischem Scheren während des Pipettierens sind entscheidend, um die langen Fragmente zu erhalten, die für die Bibliotheksvorbereitung für PacBio und Nanopore erforderlich sind.
Diagnose häufiger Qualitätsprobleme:
- Ungleichmäßige Abdeckung: GC-Bias, Fragmentierungsbias oder niedrige Bibliothekskomplexität. Lösung: Eingangsqualität neu bewerten, Bibliotheksmethode anpassen, PCR-Zyklen kontrollieren.
- Niedrige Abgleichrate: Kontamination, Abweichung des Referenzgenoms oder Probenzerfall. Lösung: Kontaminationsscreening hinzufügen, Eignung des Referenzgenoms überprüfen.
- Hohe Duplikationsrate: Niedriger DNA-Eingang, übermäßige PCR-Amplifikation oder geringe Bibliothekskomplexität. Lösung: Reduzieren Sie die PCR-Zyklen, optimieren Sie die Bibliothekskomplexität, ziehen Sie PCR-freie Protokolle in Betracht. Für Probenarten mit begrenztem Eingangsmaterial, genomische Datenanalyse kann helfen zu beurteilen, ob die Duplikationsrate innerhalb akzeptabler Grenzen für die beabsichtigte Nachweismethode liegt.
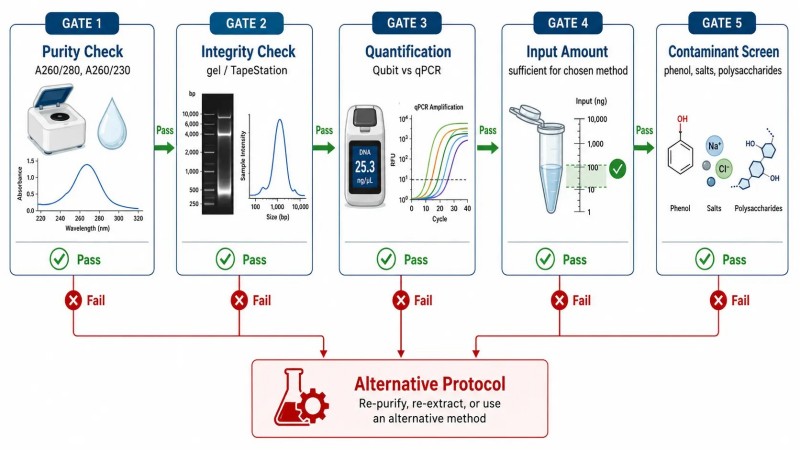 Abbildung 4. Qualitätskontrollen vor NGS — QC-Schwellenwerte und Risikobewertung
Abbildung 4. Qualitätskontrollen vor NGS — QC-Schwellenwerte und Risikobewertung
Bildunterschrift: Beispiel für Qualitätskontrollschranken für DNA- und RNA-Proben vor NGS, die quantitative QC-Schwellenwerte (A260/280, A260/230, RIN, DV200) und Risikobewertungen für häufige Qualitätsprobleme wie ungleichmäßige Abdeckung, niedrige Mapping-Rate und hohe Duplikationsrate zeigen.
Die Bibliotheksvorbereitung bestimmt die Nutzbarkeit der Daten.
Die Bibliotheksvorbereitung ist die Brücke zwischen rohen Nukleinsäuren und sequenzierungsbereiten Molekülen. Ihre Hauptfunktionen bestehen darin, DNA oder RNA in plattformkompatible Moleküle umzuwandeln, Adapter und Barcodes für die Bindung an die Flusszelle und die Identifizierung von Proben einzuführen, die Insertgröße und die Komplexität der Bibliothek zu steuern und die Stranginformation dort zu bewahren, wo dies erforderlich ist.
Schlüsselvariablen, die die Sequenzierungsausgabe beeinflussen: Die Fragmentierungsstrategie (mechanisch vs. enzymatisch vs. Tagmentation) beeinflusst die Abdeckungsbias und Reproduzierbarkeit. Die Effizienz der Adapterligatur bestimmt den Anteil der Fragmente, die sequenziert werden können. Die Anzahl der PCR-Zyklen beeinflusst direkt die Duplikationsrate – jeder zusätzliche Zyklus über 10 hinaus fügt ungefähr 5-10% mehr Duplikate hinzu. Das Größenwahlfenster steuert die Verteilung der Insertgrößen, was die Clusterdichte und Mapping-Raten beeinflusst. Die Methode zur Quantifizierung der Bibliothek (qPCR vs. Qubit vs. Bioanalyzer) muss sorgfältig ausgewählt werden – qPCR ist die genaueste Methode zur Bestimmung der sequenzierungsbereiten Konzentration.
Häufige Fehler bei der Bibliotheksvorbereitung:
- Vorausgesetzt, dass eine hohe Bibliothekskonzentration gleich hohe Bibliotheksqualität bedeutet.
- Ignorieren von Adapter-Dimer-Kontamination, die Sequenzierungslesungen verschwendet.
- Die Verwendung der totalen DNA-Quantifizierung (Qubit) allein anstelle der gleichzeitigen Messung amplifizierbarer Moleküle (qPCR)
- Übermäßige PCR-Amplifikation in Proben mit niedrigem Input, was zu hohen Duplikationsraten führt
Für eine detaillierte Diskussion über die Optimierung der Bibliotheksvorbereitung siehe die Ressource zur Vorbereitung von NGS-Bibliotheken, die Fragmentierung, Endreparatur, Adapterligierung, Amplifikation, Aufreinigung und Qualitätskontrolle im Detail abdeckt. Für Projekte, die spezialisierte Probenarten wie FFPE-Gewebe oder cfDNA betreffen, gezielte Sequenzierungsansätze erfordern häufig spezifische Bibliotheksprotokolle, die für degradierte oder material mit niedrigem Input optimiert sind.
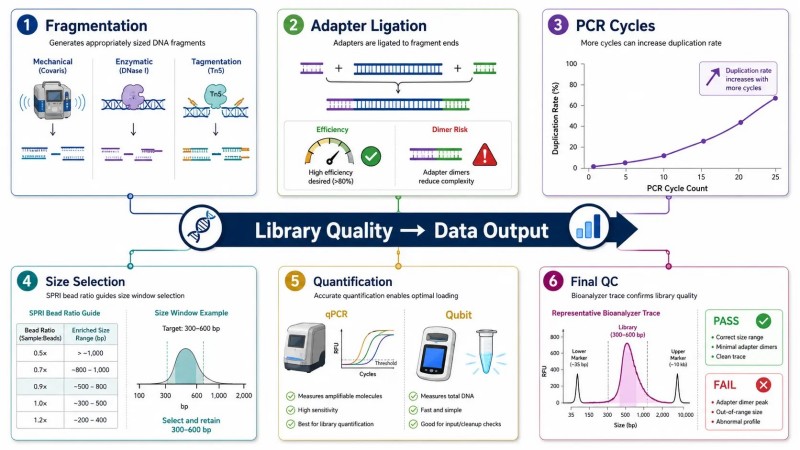 Abbildung 5. Variablen der Bibliotheksvorbereitung, die den Sequenzierungsausgang beeinflussen
Abbildung 5. Variablen der Bibliotheksvorbereitung, die den Sequenzierungsausgang beeinflussen
Beschriftung: Wichtige Variablen der Bibliotheksvorbereitung, die den NGS-Sequenzierungsausgang beeinflussen – Fragmentierungsstrategie, Effizienz der Adapterligatur, Anzahl der PCR-Zyklen, Größenwahlfenster und Quantifizierungsmethode – mit häufigen Fehlern und deren Auswirkungen auf die Datenqualität.
Lese Länge, Tiefe und Abdeckung — Drei unterschiedliche Konzepte
Diese drei Begriffe werden häufig austauschbar in Projektdiskussionen verwendet, beschreiben jedoch unterschiedliche Parameter, die jeweils die Datenqualität und die Projektkosten unabhängig beeinflussen. NGS kann auf DNA oder RNA aus nahezu jeder biologischen Quelle angewendet werden — Blut, Gewebe, Zellen, FFPE-Blöcke, Plasma (cfDNA), mikrobielle Kulturen, Umweltproben und Einzelzellen. Die entscheidende Einschränkung besteht darin, dass die Probenqualität die Anforderungen der gewählten Bibliotheksvorbereitungsmethode und der Sequenzierungsplattform erfüllen muss.
Leseumfang Die Anzahl der zusammenhängenden Basen wird pro Sequenzierlesung bestimmt. Sie beeinflusst die Genauigkeit der Ausrichtung, die Fähigkeit, sich wiederholende Regionen zu überbrücken, die Isoformauflösung in RNA-seq und die Kontinuität der Assemblierung. Längere Reads sind nicht immer besser – sie erfordern längere Laufzeiten und erzeugen weniger Gesamtreads pro Flusszelle.
Sequenzierungstiefe (oder Abdeckungsdichte) ist die durchschnittliche Anzahl der Sequenzierungen jedes Basen im Zielbereich. Sie bestimmt das Vertrauen in Variantenaufrufe – eine höhere Tiefe ermöglicht die Erkennung von Varianten mit niedrigerer Frequenz und bietet eine robustere statistische Power für die Analyse der differentiellen Expression.
Abdeckung kann sich entweder auf den Anteil des Zielgenoms beziehen, der von mindestens einem Read abgedeckt ist (Breite der Abdeckung), oder auf die Verteilung der Tiefe über das Genom (Uniformität). Projektdiskussionen sollten klarstellen, welche Bedeutung gemeint ist.
| Metrisch | Was es misst | Warum es wichtig ist | Häufige Fehlinterpretation |
|---|---|---|---|
| Leseumfang | Länge der Sequenzierungsreads | Ausrichtung, Montage, wiederholte Überbrückung | Länger bedeutet immer besser. |
| Rohdaten | Gesamtausgabe vor der Filterung | Skala ausführen | Gleichwertige nutzbare Daten |
| Saubere Daten | Gefilterte hochqualitative Reads | Nachgelagerte Eingabe | Garantiert die Qualität der Kartierung |
| Tiefe | Durchschnittliche Lesevorgänge pro Locus | Variantvertrauen | Gleich über alle Genomregionen hinweg |
| Abdeckungsuniformität | Verteilung der Tiefe | Zuverlässigkeit über Regionen hinweg | Ignoriert, wenn die mittlere Tiefe hoch aussieht. |
Projektbezogene Tiefenanforderungen: Die menschliche WGS zur Erkennung von Keimbahn-SNVs erfordert eine Standardabdeckung von 30×. Die Erkennung somatischer Mutationen bei Krebs erfordert 60-100×, um Varianten mit niedriger Frequenz zu identifizieren. Die RNA-seq-Genexpressionsanalyse erfordert 20-50 Millionen Reads pro Probe. Das 16S-Amplicon-Profiling erfordert 10.000-50.000 Reads pro Probe. Diese Zielwerte sollten als Mindestwerte verwendet werden, wobei 10-20% Übersequenzierung hinzugefügt werden sollten, um die qualitätsbedingten Variationen zwischen den Proben zu berücksichtigen.
Projektbezogene Interpretation: Für WGS sollte der Fokus auf der genomweiten Tiefe und Uniformität liegen. Ein 30× WGS-Lauf, bei dem einige Regionen mit 5× und andere mit 60× abgedeckt sind, ist nicht äquivalent zu einem Lauf, bei dem alle Regionen mit 25-35× abgedeckt sind. Metriken zur Uniformität der Abdeckung, wie der Variationskoeffizient (CV) der Tiefe über die Bins oder der Anteil des Genoms, der innerhalb von 0,2× und 2× der mittleren Tiefe liegt, bieten ein vollständigeres Qualitätsbild als die mittlere Tiefe allein. Für WES und gezielte Panels sind die wichtigsten Metriken die On-Target-Rate, die Zielabdeckung und die Uniformität der Erfassung – nicht die gesamte Datenmenge. Für RNA-seq sind die gemappten Reads pro Probe, die Abdeckung der Genkörper und die Bibliotheksstrandedness informativer als die rohe Read-Anzahl. Für Metagenomik bestimmen der Anteil der Wirtsreads, die Wiederherstellung der mikrobiellen Vielfalt und die Schwellenwerte für die Erkennung seltener Taxa, ob die Tiefe angemessen ist.
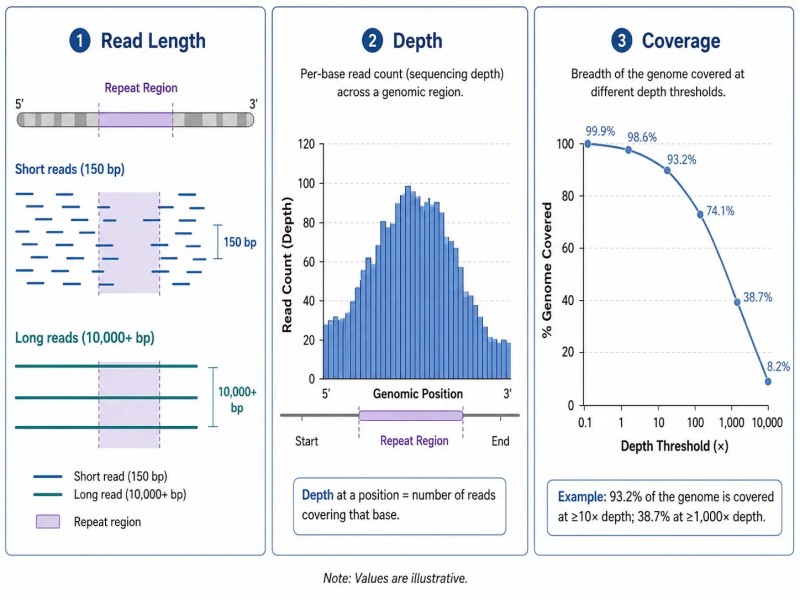 Abbildung 6. Leselänge vs. Tiefe vs. Abdeckung — drei unabhängige Metriken im Design von NGS-Projekten
Abbildung 6. Leselänge vs. Tiefe vs. Abdeckung — drei unabhängige Metriken im Design von NGS-Projekten
Beschriftung: Konzeptuelles Diagramm, das Leselänge, Sequenzierungstiefe und Abdeckung als drei unabhängige NGS-Metriken unterscheidet, mit einer Tabelle, die Definitionen, praktische Bedeutung und häufige Fehlinterpretationen für jede Metrik klarstellt.
NGS-Datenqualitätsmetriken – Worauf man in einem Sequenzierungsbericht achten sollte
Ein umfassender QC-Bericht sollte Metriken auf drei Ebenen enthalten: Sequenzierungsqualität, Ausrichtungsqualität und Bibliotheksqualität. Forscher, die einen Sequenzierungsdienstleister bewerten, sollten wissen, welche Metriken standardmäßig verwendet werden und wie man sie interpretiert.
Sequenzierungs-QC: Q20/Q30-Prozentsätze, GC-Gehaltverteilung, Adaptergehalt, Duplikationsrate und Verteilung der Lese-längen. Bei einem Standard-Illumina-Lauf sollten >85 % der Basen über Q30 für 2 × 150 bp-Läufe liegen. Die Qualitätsheatmap pro Zyklus sollte einen allmählichen Rückgang von hoher zu moderater Qualität zeigen – ein plötzlicher Abfall bei einer Zyklusnummer weist auf ein lauf-spezifisches Problem hin, das vor der Fortsetzung der Datenanalyse untersucht werden sollte.
Ausrichtungs-QC: Mapping-Rate, Prozentsatz korrekt gepaarter Reads, Verteilung der Insertgrößen, durchschnittliche Abdeckungstiefe und Abdeckungsuniformität. Niedrige Mapping-Raten (<80% für menschliche DNA) sollten untersucht werden – mögliche Ursachen sind Kontamination (bakteriell, pilzartig oder menschliche DNA durch Handhabung), Fehlanpassung des Referenzgenoms (falsche Art oder Genomversion) oder Probenabbau, der Fragmente produziert, die sich nicht eindeutig ausrichten lassen.
Bibliotheks-QC: Bibliothekskonzentration, Molarität, Größenverteilung, Adapter-Dimer-Gehalt und Schätzung der Bibliothekskomplexität. Ein Adapter-Dimer-Gehalt von über 5 % der gesamten Bibliotheksmasse verschwendet die Sequenzierungskapazität.
Projektbezogene Qualitätssicherung: Für WES oder gezielte Panels sind die On-Target-Rate und die Zielabdeckung bei bestimmten Tiefen (z. B. % der Zielbasen bei 20×, 50×, 100×) entscheidend. Whole-Exom-Sequenzierungsdienste Typischerweise werden diese Metriken als Standardlieferungen berichtet. Für RNA-seq sollten die rRNA-Rate, die Verteilung exonerer/introner/intergener Bereiche und die Abdeckung des Genkörpers berichtet werden. Für Metagenomik sollten das Niveau der Wirtskontamination, die Rate der taxonomischen Zuordnung und die Datenbankversion dokumentiert werden. Für Langzeit-Sequenzierung sind die Read N50, die Verteilung der Read-Längen, der Gesamtertrag und die Roh- vs. korrigierte Genauigkeit wichtige Metriken.
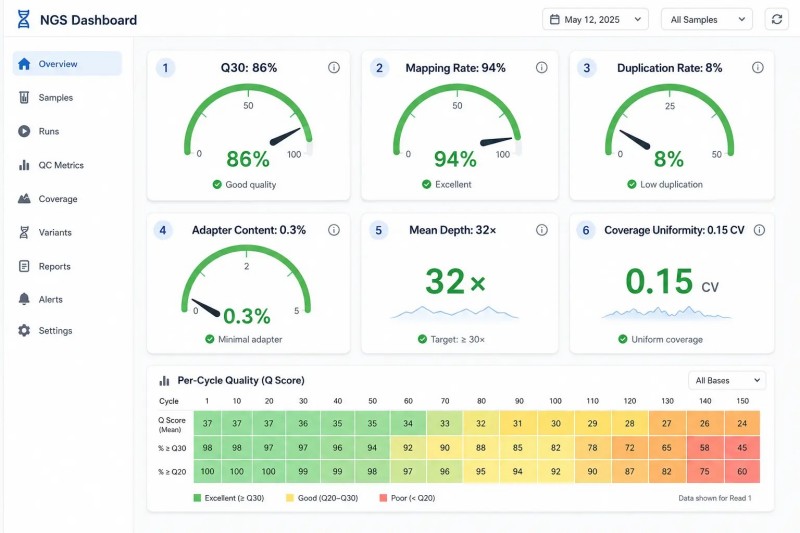 Abbildung 7. NGS QC-Dashboard — Musterbericht mit wichtigen Qualitätskennzahlen
Abbildung 7. NGS QC-Dashboard — Musterbericht mit wichtigen Qualitätskennzahlen
Überschrift: Umfassendes NGS-QC-Dashboard, das drei Ebenen von Qualitätsmetriken zeigt – Sequenzierungs-QC (Q30, GC-Gehalt, Adaptergehalt), Alignierungs-QC (Mapping-Rate, Insertgröße, Abdeckungsuniformität) und Bibliotheks-QC (Konzentration, Dimergehalt) – mit projektspezifischen Indikatoren für WES, RNA-seq, Metagenomik und Langzeitdaten.
Bioinformatische Analyse — Der Wert von NGS liegt in der Frage, nicht im Sequencer.
Das Sequenzierungsinstrument erzeugt Rohdaten. Die bioinformatische Analyse wandelt diese Daten in biologische Erkenntnisse um. Die Wahl der Analysepipeline sollte durch die Forschungsfrage bestimmt werden, nicht durch Standardeinstellungen oder Standardarbeitsabläufe.
Kernanalyse-Pipeline-Komponenten: Rohdaten-QC, Trimmen und Filtern, Ausrichtung oder Zusammenstellung, Quantifizierung oder Variantenaufruf, Annotation, statistische Analyse und biologische Interpretation. Jeder Schritt hat plattformspezifische und anwendungsspezifische Variationen, die die Ergebnisse beeinflussen.
Wesentliche Analyseunterschiede nach Projekttyp:
| NGS-Projekt | Kern-Bioinformatik-Ausgabe | Schlüsselabhängigkeit | BOFU-Frage |
|---|---|---|---|
| WGS | Variantenliste, Annotation, SV/CNV | Referenzqualität, Tiefe | Kann dieses Design meinen Zielvarianten-Typ erkennen? |
| RNA-Seq | DEG, Weg, Expressionsprofil | RNA-Qualität, Replikate | Ist das Design statistisch interpretierbar? |
| Metagenomik | Taxonomie, Funktion, Vielfalt | Datenbank, Hosterschöpfung | Können seltene Taxa oder funktionale Gene identifiziert werden? |
| Langzeitlese-Montage | Contigs, N50, BUSCO, Annotation | HMW-DNA, Abdeckung | Ist die Versammlungs-Kontinuität ausreichend für das Forschungsziel? |
Häufige Fehler in der Bioinformatik: Vorausgesetzt, dass dieselbe Pipeline für alle Projekttypen funktioniert, die Qualität und Version des Referenzgenoms ignoriert, den Einfluss der Datenbankversion auf die Annotationsergebnisse vernachlässigt, Experimente ohne biologische Replikate oder geeignete statistische Modelle entwirft und Datenvisualisierung mit biologischen Schlussfolgerungen verwechselt. Für Projekte, die maßgeschneiderte Anforderungen haben, BioinformatikanalyseDie Diskussion über Pipeline-Optionen mit dem Dienstanbieter, bevor die Sequenzierung beginnt, stellt sicher, dass das Datenformat der Ausgabe den Analyseanforderungen entspricht.
Bioinformatikanalyse-Dienstleistungen kann Pipelines bereitstellen, die auf spezifische Projekttypen zugeschnitten sind, um sicherzustellen, dass die Datenverarbeitung und -interpretation mit den Forschungszielen übereinstimmen.
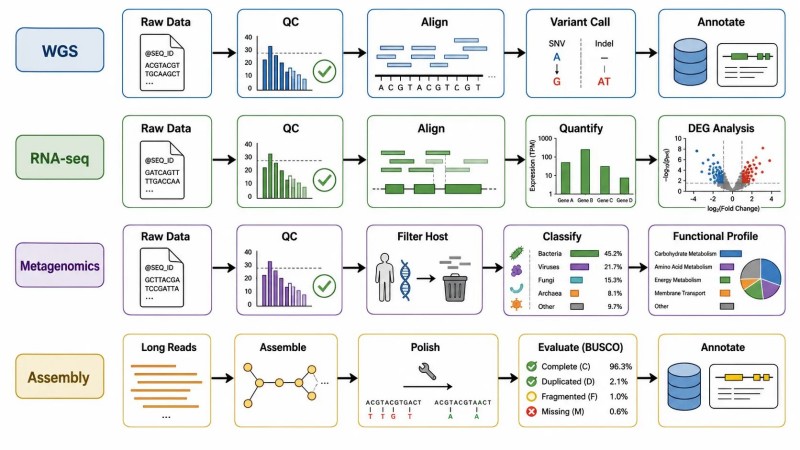 Abbildung 8. Bioinformatik-Workflow nach NGS-Projekttyp
Abbildung 8. Bioinformatik-Workflow nach NGS-Projekttyp
Bildunterschrift: Bioinformatikanalyse-Workflows für vier NGS-Projekttypen – WGS, RNA-seq, Metagenomik und Langlese-Assemblierung – die zentrale Pipeline-Komponenten, wichtige Abhängigkeiten und die BOFU (Basis of Future Use)-Frage zeigen, die vor der Auswahl der Pipeline beantwortet werden sollte.
NGS-Anwendungsdesign — WGS, WES, RNA-seq, Metagenomik und mehr
Whole Genome Sequenzierung
WGS ist geeignet für die entwicklungsgenealogische Entdeckung von Varianten, Populationsgenomik, de novo Assemblierung und vergleichende Genomik. Wichtige Entwurfsvariablen umfassen Genomgröße, Heterozygotie-Rate, Wiederholungsinhalt, erforderlichen Variantentyp und Verfügbarkeit eines Referenzgenoms. Kurzlese-WGS bei 30× ist der Standard für die Erkennung von menschlichen Keimbahn-SNVs. Langlese-WGS wird für die Assemblierung, die Erkennung struktureller Varianten und wiederholungsreiche Regionen bevorzugt. Hybride Strategien, die beide Lesetypen kombinieren, bieten das beste Gleichgewicht für eine umfassende Genomanalyse.
Für ein menschliches WGS-Projekt umfasst das Standardlieferobjekt etwa 90-100 Gb Rohdaten pro Probe bei 30×. Die bioinformatische Pipeline muss mindestens die Variantenbestimmung für SNVs, kleine Indels und Kopienzahlvarianten verarbeiten, wobei die Analyse struktureller Varianten als optionale Erweiterung vorgesehen ist. Whole-Genome-Sequenzierungsdienste kann je nach Forschungszielen für Kurzlese-, Langlese- oder Hybridansätze konfiguriert werden.
Whole Exome Sequenzierung / Targeted Sequenzierung
WES und gezielte Panels konzentrieren sich auf spezifische genomische Regionen, reduzieren die Kosten und ermöglichen eine höhere Tiefe in den Zielregionen. Wichtige Entwurfsvariablen umfassen das Design der Erfassungsregion, Erwartungen an die On-Target-Rate, Anforderungen an die Zielabdeckung, die Kompatibilität der Sonden mit der Zielart sowie GC-reiche oder sich wiederholende Zielregionen. Risiken umfassen ungleichmäßige Abdeckung über die Zielregionen, Erfassungsbias und die Unfähigkeit, Nicht-Zielregionen zu interpretieren.
Für menschliches WES umfasst ein typisches Ergebnis eine mittlere Tiefe von 100-200× in den Zielregionen, wobei mindestens 90 % der Zielbasen mit 20× oder mehr abgedeckt sein sollten. Die On-Target-Rate (Prozentsatz der Reads, die innerhalb oder in der Nähe des Capture-Designs abgebildet werden) sollte für standardmäßige Exom-Capture-Kits 60 % übersteigen.
RNA-Sequenzierung
RNA-Seq misst die Genexpression, erkennt alternatives Spleißen, identifiziert Fusionstranskripte und entdeckt neuartige Transkripte. Wichtige Variablen sind die RNA-Integrität, die Bibliotheksstrangrichtung (Erhaltung der Strangorientierung), die Poly(A)-Selektion im Vergleich zur rRNA-Depletion-Strategie und die Anzahl biologischer Replikate. Risiken umfassen RNA-Abbau, der die Quantifizierungsgenauigkeit beeinträchtigt, Batch-Effekte durch die Verarbeitung von Proben zu unterschiedlichen Zeiten und ein unzureichendes Replikatdesign für statistische Power.
Ein standardmäßiges mRNA-seq-Projekt erfordert 20-50 Millionen Reads pro Probe für die Quantifizierung auf Genebene. Für die Analyse auf Isoformenebene können mehr als 100 Millionen Reads pro Probe erforderlich sein. Es werden mindestens drei biologische Replikate pro Bedingung empfohlen, um eine zuverlässige Analyse der differentiellen Expression zu gewährleisten. RNA-Seq-Dienste Unterstützen Sie sowohl poly(A)-ausgewählte als auch rRNA-depletierte Bibliothekstypen.
Metagenomische Sequenzierung
Metagenomik profilieren die mikrobielle Gemeinschaftsstruktur, das funktionale Potenzial und die Zusammensetzung auf Stamm-Ebene. Wichtige Variablen sind der Anteil der Wirts-DNA, die mikrobielle Biomasse, die Wahl der Datenbank und die Sequenzierungstiefe. Risiken umfassen die Überwältigung der mikrobiellen Reads durch Wirtskontamination, unzureichende Tiefe für die Erkennung seltener Taxa und datenbankabhängige Annotationsverzerrungen, die je nach verwendeter Referenzsammlung variieren.
Für Shotgun-Metagenomik sind 50-100 Millionen Reads pro Probe typisch für eine umfassende funktionale Profilierung. Für 16S-Amplicon-Sequenzierung sind 10.000-50.000 Reads pro Probe ausreichend für die Analyse der Gemeinschaftszusammensetzung. Strategien zur Depletion von Wirts-DNA – einschließlich differenzieller Lyse und probe-basierter Erfassung – sollten für Mikrobiom-Proben mit niedrigem Biomasseanteil aus wirtsassoziierten Standorten in Betracht gezogen werden. 16S/ITS Amplicon-Sequenzierungsdienste Standardisierte Protokolle für die Gemeinschaftsprofilierung bereitstellen, während Shotgun-Metagenomik-Dienstleistungen höhere Auflösung für funktionale und Dehnungsanalyse anbieten.
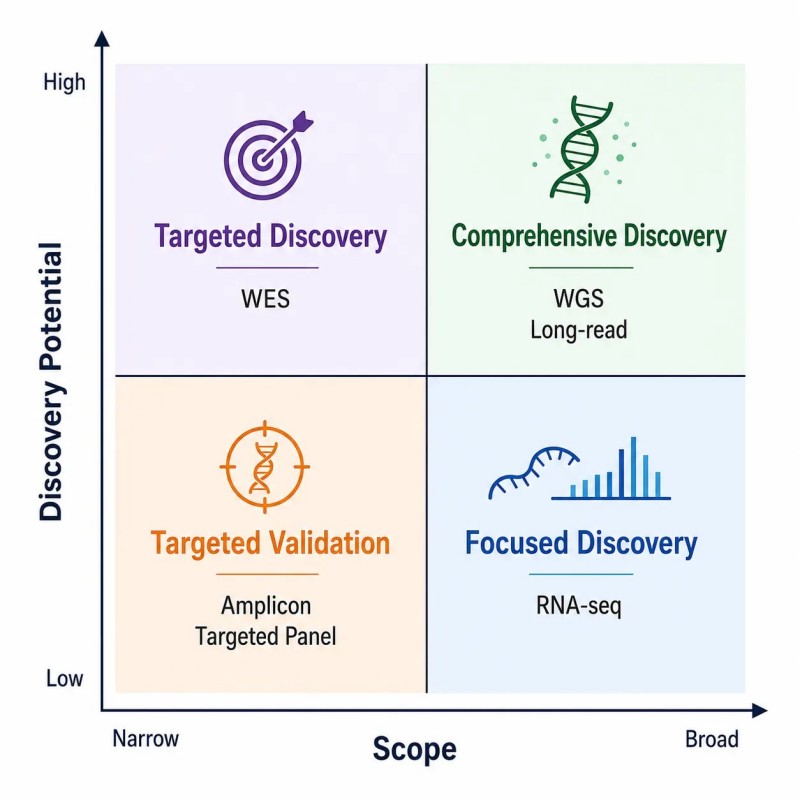 Abbildung 9. Auswahlmatrix für NGS-Anwendungen — Abstimmung von Forschungszielen auf Sequenzierungsdienste
Abbildung 9. Auswahlmatrix für NGS-Anwendungen — Abstimmung von Forschungszielen auf Sequenzierungsdienste
Beschriftung: Anwendungsauswahlmatrix für vier wichtige NGS-Ansätze – WGS, WES, RNA-seq und Metagenomik – die optimale Anwendungsfälle, Entwurfsvariablen, Risiken und empfohlene Lese-Tiefe für jeden Anwendungstyp zeigt.
Auswahl zwischen Amplicon, Panel, Exom, Genom und Transkriptom
Die Wahl zwischen diesen fünf Assaytypen hängt vom Umfang der Forschungsfrage und dem erforderlichen Entdeckungspotenzial ab.
| Option | Am besten für | Entdeckungspotenzial | Datenkomplexität | Hauptbeschränkung |
|---|---|---|---|---|
| Amplicon | Bekannte kleine Regionen, hohe Stichprobenzahlen | Niedrig | Niedrig–Mittel | Enger Umfang |
| Zielpanel | Bekannte Gen-Sets | Mittel | Mittel | Designabhängig |
| WES | Kodierungsvarianten | Mittel–Hoch | Mittel–Hoch | Fehlende nicht-kodierende Regionen |
| WGS | Genomweite Entdeckung | Hoch | Hoch | Höhere Datenlast |
| RNA-Seq | Expression / Transkriptom | Hoch für RNA-Spiegel | Hoch | RNA-Qualität empfindlich |
Käuferentscheidungslogik: Ist das Ziel bekannt oder unbekannt? Ist eine Entdeckung erforderlich oder ist eine Bestätigung ausreichend? Ist eine genomweite Abdeckung notwendig oder ist eine fokussierte Region ausreichend? Ist die Fragestellung auf DNA- oder RNA-Ebene? Unterstützen die Probenqualität und das Budget die Komplexität des gewählten Ansatzes? Die Beantwortung dieser Fragen vor der Auswahl eines Assay-Typs verhindert kostspielige Änderungen während des Projekts.
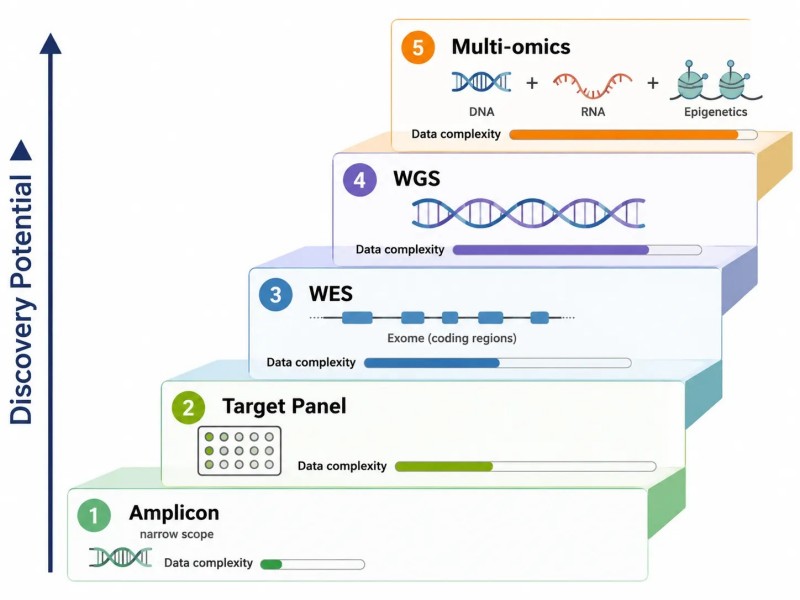 Abbildung 10. Auswahlleiter für NGS-Assays — von Amplicon bis Whole Genome
Abbildung 10. Auswahlleiter für NGS-Assays — von Amplicon bis Whole Genome
Bildunterschrift: Fünfstufige Auswahlleiter für NGS-Assays von Amplicon (geringstes Entdeckungspotenzial, engste Abdeckung) bis WGS (höchstes Entdeckungspotenzial, breiteste Abdeckung), mit Entscheidungslogik zur Auswahl des geeigneten Assays basierend auf Zielwissen und Forschungsanforderungen.
Häufige Ursachen für das Scheitern von NGS-Projekten – und wie man sie verhindern kann
Das Verständnis von Fehlermodi, bevor ein Projekt gestartet wird, ist die effektivste Präventionsstrategie. Fehler können in jeder Phase auftreten.
| Beobachtetes Problem | Mögliche Ursache | Gestaltungsbezogene Prävention |
|---|---|---|
| Niedrige nutzbare Lesevorgänge | Kontamination, Adapter-Dimere, niedrige Bibliotheksqualität | Vor-Sequenzierung QC und Reinigung |
| Ungleichmäßige Abdeckung | GC-Bias, Capture-Bias, Fragmentierungs-Bias | Anpassung der Plattform- und Bibliotheksstrategie |
| Hohe Duplizierung | Niedrige DNA-Eingabe, Über-PCR | Überwachung der Bibliothekskomplexität, PCR-freie Protokolle |
| Schwaches biologisches Signal | Schlechtes Replikatdesign, Batch-Effekte | Statistisches Design vor der Sequenzierung |
| Schlechte Annotation | Datenbankfehlanpassung, veralteter Verweis | Datenbankauswahl und Versionskontrolle |
Probeebene-Fehler: Degradation, Kontamination, unzureichende Eingabe oder schlechte Extraktionsmethode — durch strenge Qualitätskontrolle vor der Sequenzierung angehen.
Bibliotheksbezogene Fehler: Adapter-Dimer-Kontamination, niedrige Umwandlungseffizienz, Überamplifikation, verzerrte Fragmentierung — ansprechen durch Optimierung der Bibliotheksprotokolle und Einbeziehung von QC-Kontrollpunkten.
Sequenzierungsfehler: Unterladung oder Überladung der Flusszelle, Bibliotheken mit geringer Diversität, die Kalibrierungsfehler verursachen, Ungleichgewicht zwischen multiplexierten Proben, unzureichende Tiefe — dies kann durch sorgfältige Ladeberechnungen und Diversitätsplanung angegangen werden.
Datenebene-Fehler: Niedrige Mapping-Rate, schlechte Auswahl des Referenzgenoms, hohe Duplikation, Batch-Effekte, Datenbankinkonsistenzen — ansprechen durch Einbeziehung von Kontrollen und Planung der Analyse vor der Sequenzierung.
Projektentwurfsfehler: Die Forschungsfrage ist zu breit für den gewählten Ansatz, falsche Plattform oder Assay-Typ, keine biologischen Replikate, unrealistische Erwartungen an die nachgelagerten Ergebnisse – gehen Sie dies an, indem Sie das Rahmenwerk in diesem Leitfaden verwenden, bevor Sie Ressourcen binden.
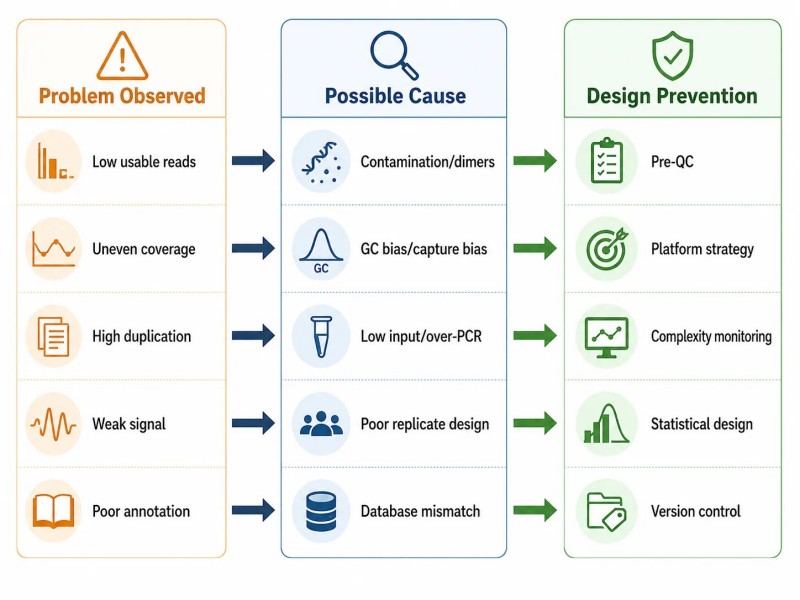 Abbildung 11. NGS-Fehlerbehebungsdiagramm — Problem, Ursache und Prävention
Abbildung 11. NGS-Fehlerbehebungsdiagramm — Problem, Ursache und Prävention
Überschrift: Umfassende NGS-Fehlerbehebungskarte, die fünf Fehlerstufen abdeckt – Probe, Bibliothek, Sequenzierung, Daten und Projektgestaltung – mit beobachteten Problemen, möglichen Ursachen und Präventionsstrategien auf Entwurfsebene für jede Kategorie.
Wie man eine Projektanfrage an einen NGS-Dienstleister vorbereitet
Eine gut vorbereitete Projektanfrage beschleunigt den Beratungsprozess und verringert das Risiko von Fehlanpassungen der Erwartungen. Die folgenden Informationen sollten vorbereitet werden, bevor Sie einen Sequenzierungsdienstleister kontaktieren.
Grundlegende Informationen, die bereitgestellt werden sollen: Art und Probenart, DNA oder RNA, Probennummer, Extraktionsmethode, Konzentration und Gesamtmenge, Integritätsdaten (RIN, DV200 oder Gelbild), Forschungsziel und erwartete Ergebnisse. Die Einbeziehung dieser Informationen in die erste Anfrage ermöglicht es dem Dienstleister, die Machbarkeit zu bewerten und eine geeignete Strategie zu empfehlen, ohne hin und her klärende Rückfragen.
Projektdesigninformationen: Ob ein Referenzgenom verfügbar ist, ob die Studie bekannte Regionen anvisiert oder Entdeckungen erfordert, ob Variantenaufruf, Expressionsprofilierung, Assemblierung oder Annotation benötigt wird, ob biologische Replikate einbezogen sind und Informationen zu Chargen.
Fragen an den Dienstleister: Welche NGS-Strategie wird empfohlen und warum? Welche QC-Metriken werden in jeder Phase berichtet? Was sind die Roh-, die bereinigten und die endgültigen Ergebnisse? Wie werden abnormale QC-Ergebnisse behandelt? Kann die bioinformatische Pipeline für das Projekt angepasst werden?
Konstruktives Stellen von Fragen: Anstelle von "Können Sie den Erfolg garantieren?" fragen Sie "Welche Stichproben- oder Entwurfsfaktoren beeinflussen die Datenqualität in diesem Projekttyp?" Anstelle von "Wie schnell können Sie liefern?" fragen Sie "Was sind die wichtigsten QC-Prüfpunkte im Projektzeitplan?"
 Abbildung 12. NGS-Projektanfrage-Checkliste — Informationen zur Vorbereitung und Fragen, die zu stellen sind
Abbildung 12. NGS-Projektanfrage-Checkliste — Informationen zur Vorbereitung und Fragen, die zu stellen sind
Überschrift: Checkliste zur Vorbereitung von Projektanfragen mit grundlegenden Informationen (Art, Probenart, Extraktionsmethode, Konzentration, Integritätsdaten), Projektdesigninformationen (Referenzgenom, Entdeckung vs. gezielt, Replikate) und konstruktiven Fragen, die an einen Sequenzierungsdienstleister gestellt werden sollten.
NGS-Strategie-Auswahl-Checkliste — Ein 10-Schritte-Rahmenwerk
- Definiere die biologische Frage.
- Identifizieren Sie, ob das Ziel DNA, RNA, epigenetisch, mikrobiell oder Einzelzelle ist.
- Bestätigen Sie die Qualität der Probe und die Durchführbarkeit der Eingabe.
- Wählen Sie den Assay-Typ: Amplicon / Panel / WES / WGS / RNA-seq / Metagenomik
- Wählen Sie die Sequenzierungsplattform: Kurzlese / Langlese / Hybrid
- Definieren Sie die Lese-Länge, Tiefe und Abdeckungs-Erwartungen.
- Bestätigen Sie die Strategie zur Bibliotheksvorbereitung.
- Definieren Sie QC-Metriken, die vor der Sequenzierung verfolgt werden sollen.
- Definieren Sie die Ergebnisse der Bioinformatik.
- Bereiten Sie Projektinformationen für die Beratung vor.
Die systematische Befolgung dieser Checkliste minimiert das Risiko kostspieliger Korrekturen während des Projekts und stellt sicher, dass die Sequenzierungsstrategie von Anfang an mit den Forschungszielen übereinstimmt.
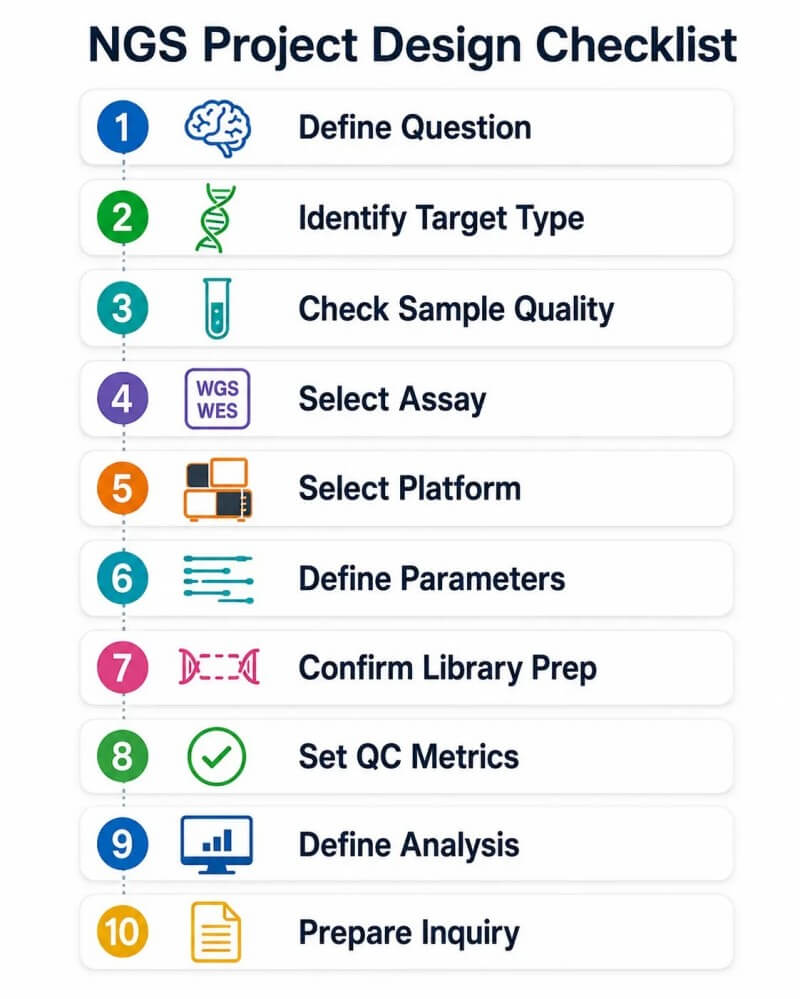 Abbildung 13. NGS-Strategie-Checkliste für Forschungsprojekte – 10-Schritte-Designrahmen
Abbildung 13. NGS-Strategie-Checkliste für Forschungsprojekte – 10-Schritte-Designrahmen
Bildunterschrift: 10-Schritte-Auswahlrahmen für NGS-Strategien von der Definition der biologischen Fragestellung über die Auswahl des Assaytyps, der Sequenzierungsplattform, der Lese- und Tiefe, der Bibliotheksstrategie, der QC-Metriken, der bioinformatischen Ergebnisse bis hin zur Vorbereitung einer Projektanfrage für eine Beratung.
Fazit — Der Wert von NGS liegt im Einklang zwischen Strategie und Fragestellung.
Die Next-Generation-Sequenzierung ist ein multivariables Projektsystem. Plattform, Bibliothek, Tiefe, Probenqualität und Analysepipeline tragen alle zur endgültigen Dateninterpretationsfähigkeit bei. Für Forscher, die NGS-Optionen bewerten, sind die wichtigsten Fragen nicht, was NGS ist, sondern welche Strategie am besten zum Forschungsziel passt, ob die Probe den gewählten Ansatz unterstützt, welche Datenmetriken im Voraus definiert werden müssen und ob die bioinformatischen Ergebnisse die ursprüngliche biologische Frage beantworten können.
Für die Planung von Forschungsprojekten können Forscher vor der Diskussion einer NGS-Strategie mit ihrem gewählten Dienstleister Informationen zu Probenart, Forschungsziel, erwarteten Analyseergebnissen und verfügbaren QC-Informationen vorbereiten.
Die erfolgreichsten NGS-Projekte sind diejenigen, bei denen das experimentelle Design von der biologischen Fragestellung geleitet wird, die Probenqualität vor Beginn der Sequenzierung bewertet wird, die Plattform und Tiefe basierend auf dem Zielvarianten-Typ und den Genommerkmalen ausgewählt werden und die bioinformatische Analyse als integraler Bestandteil des Projekts geplant wird, anstatt als nachträglicher Gedanke. Durch die Anwendung des in diesem Leitfaden beschriebenen Rahmens können Forscher das Risiko kostspieliger Korrekturen während des Projekts erheblich reduzieren und sicherstellen, dass ihre Sequenzierungsinvestition interpretierbare, veröffentlichungsreife Ergebnisse liefert.
Häufig gestellte Fragen
Was ist der Unterschied zwischen Sequenzierungstiefe und Abdeckung?
Die Tiefe bezieht sich auf die durchschnittliche Anzahl von Reads, die jede Base im Zielbereich abdecken. Die Abdeckung kann sich entweder auf den Anteil des Genoms beziehen, der von mindestens einem Read abgedeckt ist (Breite), oder auf die Gleichmäßigkeit der Tiefe im gesamten Genom. Beide Metriken sind erforderlich, um die Datenqualität zu bewerten.
Kann ich Kurz- und Langzeit-Sequenzierung in einem Projekt kombinieren?
Ja. Hybride Strategien, die lange Reads für die Kontinuität und kurze Reads für das Polieren verwenden, sind Standard für de novo Assemblierung und die Erkennung struktureller Varianten. Viele veröffentlichte Genomassemblierungen nutzen diesen kombinierten Ansatz.
Was ist die minimale DNA-Eingabe, die für NGS erforderlich ist?
Die minimale Eingabemenge variiert je nach Bibliotheksvorbereitungsverfahren: Standard-PCR-basierte Kits funktionieren mit 0,1 ng bis 1 µg, PCR-freie Kits benötigen 100 ng bis 1 µg, tagmentationsbasierte Kits arbeiten mit 1-50 ng und Ultra-Niedrig-Eingangskits können mit so wenig wie 50 pg arbeiten. Die Auswahl des geeigneten Kits für die verfügbare Eingabemenge ist entscheidend.
Wie bewerte ich die NGS-Datenqualität anhand eines Sequenzierungsberichts?
Wichtige Kennzahlen zur Überprüfung: Q30-Prozentsatz (>85% für gute Läufe), Mapping-Rate (>80% für menschliche DNA), Duplikationsrate (<15% für WGS), Adaptergehalt (<1% nach dem Trimmen) und On-Target-Rate für capture-basierte Methoden. Ein guter QC-Bericht sollte all diese Kennzahlen mit klaren Erklärungen enthalten.
Wie wähle ich zwischen WGS und WES für ein Projekt in der Humangenetik?
Wählen Sie WGS, wenn eine umfassende Variantenerkennung (einschließlich nicht-kodierender, struktureller und regulatorischer Varianten) erforderlich ist und das Budget es zulässt. Wählen Sie WES, wenn der Fokus auf kodierenden Varianten liegt und das Projekt eine höhere Tiefe in exonen Regionen zu geringeren Gesamtkosten erfordert. WES verpasst etwa 98 % des Genoms, einschließlich der meisten regulatorischen und intronischen Regionen, die zunehmend als wichtig in der Genetik komplexer Erkrankungen anerkannt werden.
Nur für Forschungszwecke.
Referenzen:
- Übersicht über den Illumina NGS-Workflow. Illumina, Inc.
- Leistungsbewertung von DNA-Sequenzierungsplattformen in der ABRF-Studie zur Next-Generation-Sequenzierung. Naturbiotechnologie. 2021;39:1348-1365.
- Die Chemie der Next-Generation-Sequenzierung. Naturwissenschaftliche Biotechnologie. 2023;41:1709-1715.
- Nanoporen-Sequenzierungstechnologie, Bioinformatik und Anwendungen. Naturbiotechnologie. 2021;39:1348-1365.
- Genaues zirkuläres Konsens-Long-Read-Sequencing. Naturwissenschaftliche Biotechnologie2019;37:1155-1162.


