
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Einzelzell- vs. Bulk-RNA-Seq: Welche Methode wählen?
Einzelzell- vs. Bulk-RNA-Seq: Welche sollte man wählen?
Der grundlegende Unterschied zwischen scRNA-seq und Bulk-RNA-seq ist die Auflösung. Bulk-RNA-seq misst die durchschnittliche Genexpression über Tausende bis Millionen von Zellen und erzeugt ein einzelnes Expressionsprofil pro Probe. scRNA-seq misst die Expression in einzelnen Zellen und generiert Tausende von Expressionsprofilen pro Probe – eines für jede erfasste Zelle.
Dieser Unterschied in der Auflösung bestimmt die Arten biologischer Fragen, die jede Methode adressieren kann. scRNA-seq ist die geeignete Wahl, wenn die Forschungsfrage Folgendes umfasst: Identifizierung und Charakterisierung seltener Zellpopulationen (<5 % der Gesamtzellen), Analyse der zellulären Heterogenität innerhalb eines Gewebes, Rekonstruktion von Entwicklungspfaden oder Verwandtschaftsbeziehungen, Charakterisierung des Tumormikroumfelds auf Einzelzellebene oder Identifizierung zellspezifischer Reaktionen auf Behandlungen. Bulk RNA-seq ist die geeignete Wahl, wenn die Frage den Vergleich durchschnittlicher Expressionsniveaus über Bedingungen in homogenen Zellpopulationen umfasst oder wenn die Forschung einen hohen Durchsatz über viele Proben erfordert und eine Zelltypauflösung nicht erforderlich ist.
Die Entscheidung zwischen den beiden Methoden hängt auch von Kosten und Komplexität ab. Ein Standard-scRNA-seq-Experiment kostet 5-10× mehr pro Probe als Bulk-RNA-seq, und die Datenanalyse erfordert spezialisierte Computerwerkzeuge sowie erheblich mehr Speicher und Arbeitsspeicher. Für Projekte, bei denen die Zelltypauflösung nicht entscheidend ist, ist Bulk-RNA-seq die effizientere Wahl. Für Projekte, bei denen die zelluläre Heterogenität die zentrale Frage ist, liefert scRNA-seq Informationen, auf die Bulk-Methoden keinen Zugriff haben.
Praktische Anleitung – wann jede Methode verwendet werden sollteFür einen Forscher, der die Immunantworten auf einen Impfstoff untersucht, bietet die Bulk-RNA-Sequenzierung von sortierten T-Zellen aus Blutproben einen kosteneffizienten Ansatz zur Messung transkriptioneller Veränderungen in einer definierten Zellpopulation. Für einen Forscher, der die Tumorheterogenität in einer soliden Tumorbiopsie untersucht, ist die Einzelzell-RNA-Sequenzierung (scRNA-seq) die einzige Methode, die das Gemisch aus Krebszellen, stromalen Zellen, Immunzellen und Endothelzellen im Sample auflösen kann. Die Wahl sollte davon abhängen, ob die biologische Fragestellung eine Einzelzellauflösung erfordert oder mit Durchschnittswerten auf Populationsebene beantwortet werden kann.
 Abbildung 1. Vergleich von scRNA-seq und Bulk-RNA-seq — Auflösung, Kosten und Datenkomplexität
Abbildung 1. Vergleich von scRNA-seq und Bulk-RNA-seq — Auflösung, Kosten und Datenkomplexität
Vergleichende Übersicht über Einzelzell- und Bulk-RNA-Sequenzierung, die Unterschiede in der Auflösung (einzelne Zellen vs. Gewebe-Durchschnittswerte), Kosten pro Probe, Datenkomplexität und geeignete Forschungsanwendungen für jede Methode zeigt.
Experimentelle Entwurfsfaktoren, die die Datenqualität bestimmen
Die Qualität von scRNA-seq-Daten wird durch Entscheidungen bestimmt, die getroffen werden, bevor die Sequenzierung beginnt. Mehrere Faktoren beeinflussen direkt die Anzahl und Qualität der gewonnenen Zellen sowie die Zuverlässigkeit der nachgelagerten Analyse.
Probenvorbereitung und ZellauflösungDie Qualität von Einzelzell-Daten hängt entscheidend von der Qualität der Einzelzell-Suspension ab. Gewebezerfallsmethoden müssen die Zellviabilität erhalten, während sie einzelne Zellen freisetzen. Die enzymatischen Verdauungszeiten und -temperaturen sollten für jeden Gewebetyp optimiert werden – Überverdauung verursacht Stressreaktionen, die die Genexpression verändern, während Unterverdauung Aggregate und Doppelzellen produziert. Für gefrorene oder fixierte Proben sind spezifische Protokolle erforderlich, um intakte Zellkerne oder RNA zu gewinnen. Für herausfordernde Probenarten wie Fettgewebe, Knochen oder Pflanzenmaterial wurden spezialisierte Zerfallprotokolle entwickelt, die getestet werden sollten, bevor man sich auf ein großangelegtes Experiment festlegt. Einzelzell-Sequenzierungsdienste kann Protokollempfehlungen basierend auf Probenart und Forschungszielen bereitstellen.
ZielzellenzahlDie Anzahl der zu erfassenden Zellen hängt von der erwarteten Häufigkeit der Zellpopulation von Interesse ab. Um seltene Zelltypen (<1% der Gesamtzellen) zu identifizieren, wird empfohlen, 10.000-20.000 Zellen pro Probe anzustreben, um eine ausreichende Repräsentation sicherzustellen. Für die Charakterisierung der Hauptzelltypen in einem Gewebe können 3.000-5.000 Zellen ausreichend sein. Multiplexing-Strategien, die Zell-Hashing oder lipidmarkierte Indizes verwenden, können den Durchsatz erhöhen und die Kosten pro Probe senken, indem mehrere Proben in einer einzigen Erfassungsreaktion verarbeitet werden. Der Nachteil ist eine erhöhte technische Komplexität beim Demultiplexen und potenzielle Kreuzkontamination zwischen den Proben.
SequenzierungstiefeFür die Analyse auf Genebene (Erkennung, welche Gene exprimiert werden und ihre relative Häufigkeit) sind typischerweise 20.000-50.000 Reads pro Zelle ausreichend. Für die Analyse auf Isoformebene oder zur Erkennung schwach exprimierter Gene können 50.000-100.000 Reads pro Zelle erforderlich sein. Die Gesamtkosten für die Sequenzierung werden ermittelt, indem die Reads pro Zelle mit der Anzahl der Zellen multipliziert werden – ein Experiment mit 10.000 Zellen bei 50.000 Reads pro Zelle erfordert 500 Millionen Reads, was vergleichbar ist mit den Sequenzierungskosten eines Bulk-RNA-seq-Projekts mit 15-20 Proben.
PlattformauswahlDie 10x Genomics Chromium-Plattform ist das am weitesten verbreitete System, das 3'-Genexpression, 5'-Immunprofiling und multi-omische Ausgaben (CITE-seq, Feature Barcode) unterstützt. Ihre breite Akzeptanz bedeutet umfangreiche Unterstützung durch die Gemeinschaft, validierte Protokolle und Kompatibilität mit den meisten nachgelagerten Analysewerkzeugen. Plattenbasierte Methoden wie SMART-seq bieten eine vollständige Transkriptabdeckung und höhere Sensitivität pro Zelle, was sie für die Isoformdetektion und Studien, die eine vollständige Transkriptabdeckung erfordern, geeignet macht, aber der Durchsatz ist auf Hunderte von Zellen anstelle von Tausenden begrenzt. Die Wahl zwischen tropfenbasierten und plattenbasierten Methoden sollte durch die erforderliche Zellzahl geleitet werden: tropfenbasiert für Tausende von Zellen mit niedrigerer Auflösung pro Zelle, plattenbasiert für Hunderte von Zellen mit höherer Auflösung pro Zelle. Einzelzell-Sequenzierungsdienste kann je nach Projektanforderungen sowohl tropfenbasierte als auch plattenbasierte Plattformen unterstützen.
Biologische ReplikateMindestens drei biologische Replikate pro Bedingung werden für scRNA-seq-Experimente empfohlen, um die biologische Variabilität zwischen den Proben zu berücksichtigen. Das Poolen von Proben vor der Sequenzierung mit Zell-Hashing kann den Durchsatz erhöhen und gleichzeitig die Replikatinformationen beibehalten. Im Gegensatz zu Bulk-RNA-seq, bei dem jede Probe ein Expressionsprofil erzeugt, produziert scRNA-seq Tausende von Profilen pro Probe, was ein falsches Gefühl für die statistische Power erzeugen kann – selbst bei Tausenden von Zellen können Ergebnisse aus einer einzigen Probe nicht verallgemeinert werden, da sie möglicherweise probe-spezifische und nicht bedingungsspezifische Effekte widerspiegeln.
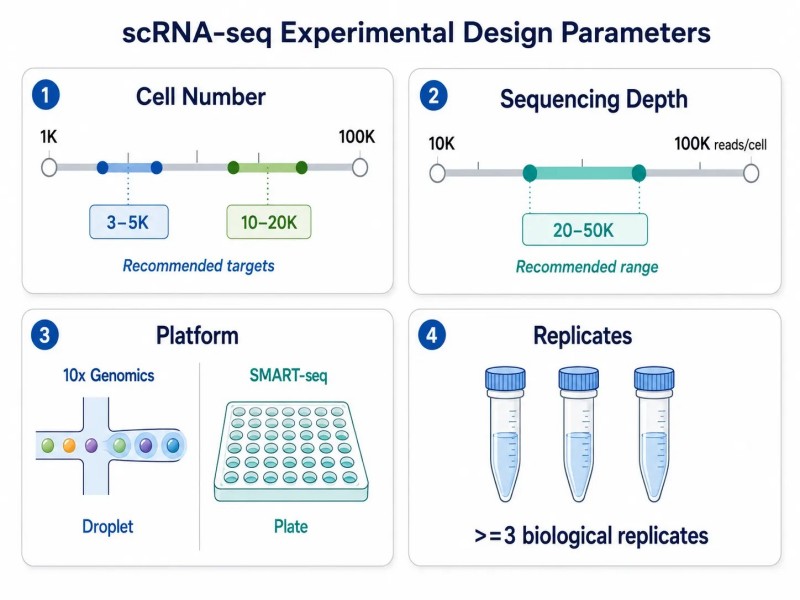 Abbildung 2. scRNA-seq Experimentdesign — Schlüsselfaktoren und empfohlene Bereiche
Abbildung 2. scRNA-seq Experimentdesign — Schlüsselfaktoren und empfohlene Bereiche
Bildunterschrift: Wichtige experimentelle Entwurfsparameter für scRNA-seq mit empfohlenen Bereichen für die Probenvorbereitung, Zielzellzahl, Sequenzierungstiefe, Plattformauswahl (10x vs. SMART-seq) und biologische Replikate.
Die Standard scRNA-Seq Analyse-Pipeline
Die standardmäßige scRNA-seq-Analysepipeline folgt einer strukturierten Abfolge von sechs Phasen, die jeweils spezifische Werkzeugauswahlen und Parameterentscheidungen umfassen: Qualitätskontrolle und Zellfilterung, Normalisierung, Batch-Korrektur, Dimensionsreduktion und Clusterbildung, Zelltypannotation und nachgelagerte biologische Analyse. Jede Phase produziert Zwischenoutputs, die vor dem Übergang zur nächsten Phase überprüft werden sollten – das Überspringen dieses Inspektionsschrittes ist eine häufige Ursache für schlechte Endergebnisse.
wird häufig Seurat empfohlen, da es eine benutzerfreundlichere Oberfläche und umfangreiche Dokumentation bietet. Scanpy hingegen kann für erfahrene Programmierer, die maßgeschneiderte Analysen durchführen möchten, vorteilhafter sein. Beide Tools haben ihre eigenen Stärken und Schwächen, und die Wahl hängt letztlich von den spezifischen Anforderungen des Projekts und den Fähigkeiten des Forschungsteams ab. Bioinformatik-Dienstleistungen kann standardisierte scRNA-seq-Analyse-Pipelines bereitstellen, die QC, Normalisierung, Integration und Annotation mit dokumentierten Parameter-Einstellungen behandeln. Genomdatenanalyse-Dienste kann auch benutzerdefinierte nachgelagerte Analysen unterstützen, einschließlich Pseudotime- und Zell-Zell-Kommunikationsstudien.
QC und Zellfilterung — Quantifizierbare Schwellenwerte
Die Qualitätskontrolle in der scRNA-seq umfasst das Filtern von Zellen, die wahrscheinlich technische Artefakte und nicht echte biologische Signale sind. Drei Metriken werden als Standard-QC-Filter verwendet:
- Einzigartige Genanzahl (nFeature_RNA)Zellen mit weniger als 200-500 erkannten Genen sind typischerweise leere Tropfen oder tote Zellen. Zellen mit mehr als 5.000-7.500 Genen können Doppelzellen sein (zwei Zellen, die in einem Tropfen erfasst wurden). Die Schwellenwerte sollten je nach Zelltyp angepasst werden – größere Zellen exprimieren natürlicherweise mehr Gene als kleinere Zellen.
- Mitochondriale Leseprozentsatz (percent.mt)Ein hoher mitochondrialer Gehalt (>15-20%) weist auf Zellen mit beschädigten Membranen hin, die zytoplasmatische RNA verloren haben. Diese Zellen sollten entfernt werden, da ihre Expressionsprofile von mitochondrialen Transkripten dominiert werden und nicht das wahre Transkriptom der Zelle widerspiegeln.
- DoppelterkennungDie computergestützte Doppelterkennung mit Tools wie DoubletFinder, scDblFinder oder scrublet identifiziert Zellen, deren Expressionsprofile einer Mischung aus zwei verschiedenen Zelltypen ähneln. Eine Doppeltrate von 3-8 % ist typisch für Standard-10x-Erfassungen. Höhere Raten deuten auf eine suboptimale Zellbeladung hin.
Diese Schwellenwerte sollten vor und nach der Filterung mithilfe von Violin-Plots und Streudiagrammen visualisiert werden. Die Entscheidung zur Filterung sollte auf der Verteilung dieser Metriken über alle Zellen basieren, nicht auf willkürlich festgelegten Schwellenwerten. Eine Zellpopulation mit natürlicherweise hohem mitochondrialen Gehalt (z. B. Nieren- oder Leberzellen) sollte andere Filterungsschwellen haben als Immunzellen. Nach der Filterung sollte der Prozentsatz der erhaltenen Zellen als Teil des Analyseberichts dokumentiert werden – das Entfernen von mehr als 30-40 % der Zellen erfordert eine Überprüfung des Dissoziationsprotokolls oder der Probenqualität.
Entfernung leerer TropfenEin kritischer Vorverarbeitungsschritt, der spezifisch für tropfenbasierte scRNA-seq ist, besteht darin, leere Tropfen (die Umgebungs-RNA, aber keine Zellen enthalten) von echten Zellen zu unterscheiden. Die Standardfilterung von CellRanger verwendet einen UMI-Zählgrenzwert, aber ausgefeiltere Methoden wie EmptyDrops (DropletUtils-Paket) verwenden einen statistischen Test, um Barcodes mit Expressionsprofilen zu identifizieren, die sich vom Hintergrund der Umgebungs-RNA unterscheiden. Die Verwendung von EmptyDrops anstelle eines festen UMI-Grenzwerts ermöglicht die Wiedergewinnung kleiner Zellen mit niedrigem RNA-Gehalt und entfernt die Kontamination mit Hintergrund-RNA aus den verbleibenden Zellen.
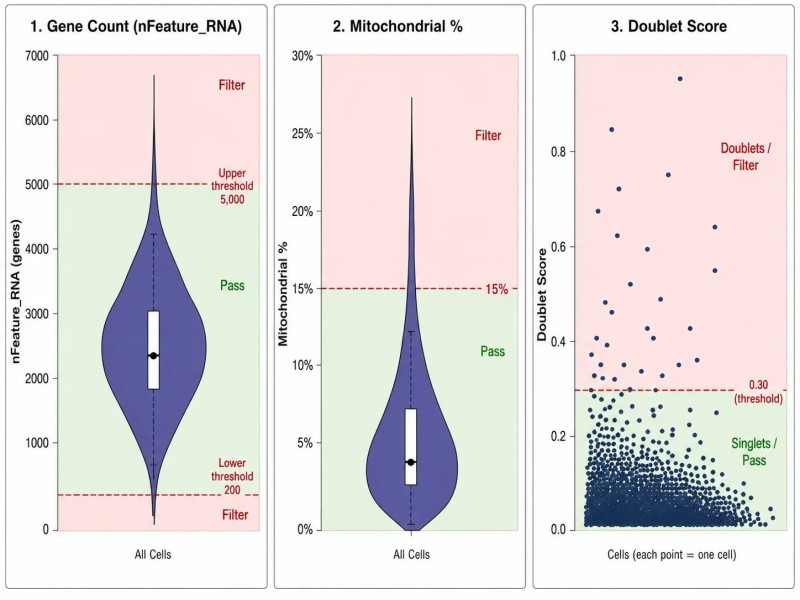 Abbildung 3. scRNA-seq QC-Filtergrenzen — Genanzahl, mitochondrialer Prozentsatz und Doppelterkennung
Abbildung 3. scRNA-seq QC-Filtergrenzen — Genanzahl, mitochondrialer Prozentsatz und Doppelterkennung
Beschriftung: Qualitätskontrollschwellen für scRNA-seq, die Violin-Diagramme und Streudiagramme für die Anzahl einzigartiger Gene (nFeature_RNA), den Prozentsatz mitochondrialer Reads (percent.mt) und die Erkennung computergestützter Doppelten zeigen, mit empfohlenen Filterbereichen für jede Kennzahl.
Normalisierung und Batch-Korrektur — Die richtige Methode wählen
Die Normalisierung in der scRNA-seq muss sowohl technische Variationen (Unterschiede in der Erfassungs-effizienz, Sequenzierungstiefe zwischen Zellen) als auch biologische Variationen (Unterschiede in Zellgröße und RNA-Gehalt) berücksichtigen.
NormalisierungsmethodenSCTransform (Seurat) ist die am weitesten verbreitete Methode zur Normalisierung von scRNA-seq-Daten. Es modelliert UMI-Zählungen mithilfe einer regularisierten negativen binomialen Regression, die die Sequenzierungstiefe berücksichtigt und gleichzeitig biologische Variation bewahrt. SCTransform identifiziert die technischen Quellen der Variation effektiver als die Log-Normalisierung und erzeugt Residuen, die bereit für die nachgelagerte Analyse sind. Es identifiziert auch hochvariable Gene als Teil des Normalisierungsprozesses, wodurch der Bedarf an einem separaten HVG-Auswahlschritt entfällt. Der Nachteil ist die Rechenkosten — SCTransform ist langsamer als die Log-Normalisierung und kann 16-32 GB RAM für Datensätze erfordern, die 20.000 Zellen überschreiten.
Die Scran-Methode verwendet eine auf Pooling basierende Strategie zur Schätzung von Größenfaktoren für Zellgruppen und erzeugt normalisierte Zählwerte, die zwischen Zellen vergleichbar sind. Sie ist rechnerisch effizient und funktioniert gut für Datensätze mit ausgewogenen Zelltypanteilen. Die Log-Normalisierung (log(CPM + 1)) ist der einfachste Ansatz, berücksichtigt jedoch nicht die Beziehung zwischen Sequenzierungstiefe und der Variabilität der Genexpression, die in scRNA-seq-Daten inhärent ist, weshalb sie als die am wenigsten empfohlene Methode gilt.
Batch-KorrekturWenn mehrere Proben in verschiedenen Erfassungsreaktionen oder Sequenzierläufen verarbeitet werden, sind Batch-Effekte unvermeidlich. Harmony ist eine schnelle, effektive Methode, die Batch-Effekte im PCA-Einbettungsraum korrigiert. Sie funktioniert gut für die meisten Datensätze und ist robust gegenüber Unterschieden in der Zelltypzusammensetzung zwischen den Batches, was sie zu einer guten Standardwahl für die Integration mehrerer Proben macht. Der Seurat-Integrationsworkflow (FindIntegrationAnchors + IntegrateData) verwendet die kanonische Korrelationsanalyse (CCA), um gemeinsame Zellzustände über Batches hinweg zu identifizieren, und ist die empfohlene Methode, wenn starke Batch-Effekte zu erwarten sind oder wenn Daten von verschiedenen Plattformen integriert werden. MNN (mutual nearest neighbors) korrigiert Batch-Effekte auf der Expressionsebene und ist geeignet für Datensätze, bei denen die gleichen Zelltypen in allen Batches erwartet werden.
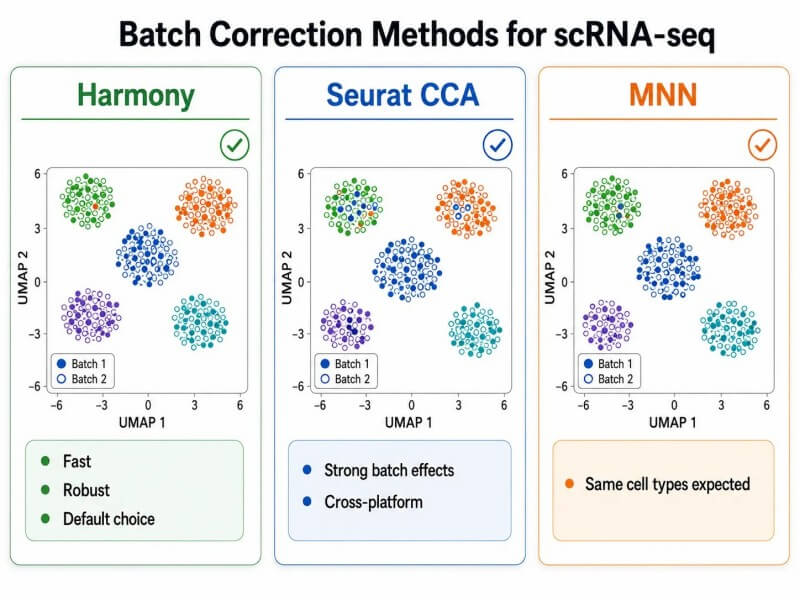 Abbildung 4. Batch-Korrekturmethoden für scRNA-seq – Vergleich von Harmony, Seurat CCA und MNN
Abbildung 4. Batch-Korrekturmethoden für scRNA-seq – Vergleich von Harmony, Seurat CCA und MNN
Überschrift: Vergleich von drei Methoden zur Batch-Korrektur für scRNA-seq—Harmony, Seurat CCA-Integration und MNN—die ihre Korrekturstrategien, rechnerischen Anforderungen und am besten geeigneten Anwendungsfälle für die Datensatzintegration zeigen.
Dimensionsreduktion und Clustering
Nach der Normalisierung und Batch-Korrektur wird die hochdimensionale Genexpressionsmatrix auf eine niederdimensionale Darstellung reduziert, um sie zu visualisieren und zu clustern.
Hauptkomponentenanalyse (PCA)PCA ist der standardmäßige erste Schritt bei der Dimensionsreduktion. Für die meisten scRNA-seq-Datensätze erfassen 20-50 Hauptkomponenten die bedeutende biologische Variation. Das Ellenbogen-Diagramm (erklärte Varianz pro PC) wird verwendet, um die optimale Anzahl an PCs zu bestimmen – der Punkt, an dem die Kurve abflacht, zeigt den Cutoff an, jenseits dessen die Komponenten hauptsächlich Rauschen erfassen. Zu wenige PCs auszuwählen, verwirft biologische Variation, die relevant ist, um ähnliche Zelltypen zu unterscheiden; zu viele auszuwählen, führt zu Rauschen, das die Clusterstruktur verschleiern kann.
UMAP-VisualisierungUMAP bietet eine 2D-Darstellung der zellulären Landschaft, die sowohl lokale als auch globale Strukturen bewahrt. Es hat t-SNE weitgehend für die Visualisierung von scRNA-seq ersetzt, aufgrund seiner Geschwindigkeit und der besseren Erhaltung globaler Beziehungen zwischen Zellclustern.
ClusterbildungDie Louvain- und Leiden-Algorithmen sind die Standardmethoden zur Identifizierung von Zellclustern. Leiden wird gegenüber Louvain bevorzugt, da es gut verbundene Cluster garantiert und weniger wahrscheinlich getrennte Gemeinschaften erzeugt. Der Auflösungsparameter steuert die Granularität der Clusterbildung – eine höhere Auflösung erzeugt mehr Cluster, die möglicherweise verschiedene Zellsubtypen repräsentieren, kann jedoch auch zu einer übermäßigen Aufspaltung kontinuierlicher Zellpopulationen führen. Ein typischer Arbeitsablauf testet Auflösungen von 0,2 bis 1,2 und wählt die Auflösung aus, die biologisch interpretierbare Cluster ohne übermäßige Fragmentierung erzeugt.
Cluster-Marker-IdentifikationSobald die Cluster definiert sind, werden Marker-Gene für jedes Cluster identifiziert, indem das Expressionsprofil jedes Clusters mit dem aller anderen verglichen wird. Die Seurat-Funktion FindAllMarkers mit dem Wilcoxon-Rangsummentest ist die Standardmethode. Die Ausgabe ist eine Liste von Genen, die in jedem Cluster hochreguliert sind, sortiert nach durchschnittlichem log2-Faltenwechsel oder angepasstem p-Wert. Diese Marker-Gene werden zur Annotation von Zelltypen verwendet und sollten im Kontext bekannter Biologie interpretiert werden – ein Cluster, das T-Zell-Marker (CD3D, CD8A) exprimiert, ist wahrscheinlich eine T-Zell-Population, während eines, das B-Zell-Marker (CD79A, MS4A1) exprimiert, wahrscheinlich eine B-Zell-Population ist.
Zelltypannotation — Manuell vs. Automatisiert
Die Zelltypannotation ist der Schritt, der Clusteridentitäten in biologische Bedeutung übersetzt. Es stehen zwei Ansätze zur Verfügung, die unterschiedliche Vor- und Nachteile haben.
Manuelle AnnotationBekannte Marker-Gene für jeden erwarteten Zelltyp werden verwendet, um Cluster basierend auf ihren Expressionsprofilen zu kennzeichnen. Die manuelle Annotation ist der Goldstandard für Genauigkeit, ist jedoch zeitaufwändig und erfordert Fachkenntnisse über das untersuchte Gewebe oder den Zelltyp. Sie wird für Projekte empfohlen, bei denen die Genauigkeit der Annotation entscheidend ist, wie z. B. klinische Studien oder Projekte, die sich auf die Identifizierung neuer Zellsubtypen konzentrieren.
Automatisierte AnnotationWerkzeuge wie SingleR, CellTypist und ScType vergleichen das Expressionsprofil jeder Zelle mit Referenzdatensätzen, um automatisch Zelltypbezeichnungen zuzuweisen. Die automatisierte Annotation ist schnell und reproduzierbar, hängt jedoch stark von der Qualität und Relevanz des Referenzdatensatzes ab. Wenn die Referenz keine Zelltypen enthält, die im Abfragedatensatz vorhanden sind, werden diese Zellen falsch klassifiziert oder nicht zugewiesen. Eine praktische Strategie besteht darin, die automatisierte Annotation als ersten Schritt zu verwenden und die Ergebnisse anschließend durch manuelle Inspektion von Marker-Genen zu validieren oder zu verfeinern.
Für Projekte, die eine validierte Zelltypannotation mit geeigneten Qualitätskontrollen erfordern, Bioinformatikanalyse-Dienstleistungen kann sowohl automatisierte als auch manuelle Annotierungsstrategien mit dokumentierten Marker-Gen-Sets und Kreuzvalidierungsschritten bereitstellen.
Downstream-Analyse-Toolkit
Sobald Zelltypen identifiziert sind, können je nach Forschungsfrage eine Reihe von nachgelagerten Analysen durchgeführt werden.
- Differenzielle Ausdrucksanalyse (DE)Identifiziert Gene, die zwischen Zelltypen oder zwischen Bedingungen innerhalb eines Zelltyps unterschiedlich exprimiert sind. Der Wilcoxon-Rangsummentest (Standard von Seurat) oder MAST sind gängige Methoden. Pseudobulk-Ansätze, die Zählungen nach Probe und Zelltyp aggregieren, bevor sie Bulk-DE-Methoden (DESeq2, edgeR) anwenden, liefern konservativere und reproduzierbarere Ergebnisse.
- Gen-Set-AnreicherungsanalyseÜberprüft, ob DE-Gene in spezifischen Wegen oder funktionalen Kategorien angereichert sind. GSEA oder Überrepräsentationsanalyse unter Verwendung von GO-, KEGG- oder Reactome-Datenbanken.
- Pseudotime-TrajektorienanalyseRekonstruiert Entwicklungs- oder Differenzierungstrajektorien aus scRNA-seq-Daten, indem Zellen entlang eines kontinuierlichen Pfades basierend auf transkriptioneller Ähnlichkeit angeordnet werden. Monocle 3 und Slingshot sind Standardwerkzeuge für die Trajektorieninferenz. scVelo verwendet RNA-Geschwindigkeit, um zukünftige Zellzustände und Richtungen abzuleiten.
- Zell-Zell-KommunikationsanalyseVorhersage von Ligand-Rezeptor-Interaktionen zwischen Zelltypen unter Verwendung von Datenbanken wie CellChat, NicheNet oder SingleCellSignalR.
- Inferenz von Kopienzahlvariationen (CNV)Identifiziert großflächige chromosomale Veränderungen aus scRNA-seq-Daten mithilfe von Werkzeugen wie InferCNV, insbesondere relevant in der Krebsforschung.
 Abbildung 5. Häufige Fallen bei scRNA-seq – Probleme, Ursachen und Lösungen
Abbildung 5. Häufige Fallen bei scRNA-seq – Probleme, Ursachen und Lösungen
Zusammenfassung häufiger Fallstricke bei der Analyse von scRNA-seq, einschließlich niedriger Zellrückgewinnung, hoher Doppelraten, dominierender Batch-Effekte bei der Clusterbildung, schwer interpretierbarer Cluster durch Über-Clusterung und Unsicherheit bei der Annotation aufgrund von Referenzabweichungen.
Rechen- und Speicheranforderungen für scRNA-Seq
scRNA-seq-Projekte erzeugen erheblich mehr Daten und erfordern mehr Rechenressourcen als Bulk-RNA-seq-Projekte mit vergleichbarer Stichprobengröße.
- Rohdaten pro 10x-ErfassungEin standardmäßiger 10x-Lauf, der 10.000 Zellen mit 50.000 Reads pro Zelle anvisiert, produziert ungefähr 500 Millionen Reads und erzeugt 30-50 GB FASTQ-Daten pro Probe.
- SpeicheranforderungenFür ein Projekt mit 10 Proben planen Sie ungefähr 300-500 GB an Rohdaten sowie 100-200 GB für ausgerichtete und verarbeitete Dateien. Insgesamt: 500-700 GB.
- SpeicheranforderungenDie Seurat- und Scanpy-Analyse von 10.000 Zellen erfordert 16-32 GB RAM. Für Datensätze mit mehr als 50.000 Zellen werden 64-128 GB empfohlen.
- BerechnungszeitEin standardmäßiger Seurat-Workflow für 10.000 Zellen dauert 2-4 Stunden. Für 100.000 Zellen planen Sie 12-24 Stunden ein. Scanpy-Workflows sind im Allgemeinen schneller und speichereffizienter für große Datensätze.
Neue Richtungen — Multi-Omik und räumliche Integration
Die Einzelzelltechnologie entwickelt sich über die Transkriptomik hinaus, um mehrere molekulare Schichten aus derselben Zelle zu erfassen. CITE-seq misst gleichzeitig die Genexpression und die Oberflächenproteinmenge mithilfe von oligonukleotidkonjugierten Antikörpern. scATAC-seq profiliert die Chromatinzugänglichkeit mit Einzelzellauflösung. Einzelzell-Multi-Omics-Plattformen (10x Multiome) erfassen die RNA-Expression und ATAC-seq aus derselben Zelle in einer einzigen Reaktion.
die Integration von scRNA-seq mit räumlicher Transkriptomik ist eines der aktivsten Bereiche der methodischen Entwicklung. Plattformen für räumliche Transkriptomik (10x Visium, Slide-seq, MERFISH, Xenium) kartieren die Genexpression auf Gewebelokationen und bieten räumlichen Kontext für die Zelltypen, die durch scRNA-seq identifiziert wurden. Computermethoden wie RCTD, Cell2location und SpaGCN ermöglichen die Integration von scRNA-seq Referenzdaten mit räumlichen Daten, um die räumliche Organisation von Zelltypen abzuleiten. Für Forschungsteams, die planen, diese Ansätze zu integrieren, Multi-Omics-Analyse-Dienste kann die Datenintegration über transkriptomische, epigenomische und räumliche Modalitäten unterstützen.
Häufige Fallstricke bei scRNA-Seq und wie man sie vermeidet
| Beobachtetes Problem | Ursache | Prävention |
|---|---|---|
| Niedrige Zellrückgewinnung | Schlechte Dissoziation, geringe Lebensfähigkeit, suboptimale Beladung | Optimieren Sie das Dissoziationsprotokoll; bewerten Sie die Lebensfähigkeit vor dem Laden. |
| Hohe Doppelrate (>10%) | Übermäßige Zellbeladungskonzentration | Berechnen Sie die Lade sorgfältig; verwenden Sie die Erkennung von Rechen-Doppeletiketten. |
| Batch-Effekte dominieren das Clustering | Verschiedene Chargen nicht ausgewogen | Verwenden Sie Zell-Hashing; fügen Sie eine Batch-Korrektur in die Pipeline ein. |
| Uninterpretable Clusterungen | Überclusterung; leere Tropfen eingeschlossen | Testen Sie mehrere Auflösungen; filtern Sie leere Tropfen rigoros. |
| Annotierungsunsicherheit | Fehlende Marker-Gene; Referenzübereinstimmung fehlt | Verwenden Sie mehrere Annotationsstrategien; validieren Sie mit unabhängigen Markern. |
Häufig gestellte Fragen
Wie viele Zellen benötige ich für scRNA-seq?
Zur Charakterisierung der Hauptzelltypen in einem Gewebe sind typischerweise 3.000-5.000 Zellen pro Probe ausreichend. Um seltene Zellpopulationen (<1% der Gesamtzellen) nachzuweisen, zielen Sie auf 10.000-20.000 Zellen ab. Die erforderliche Anzahl hängt von der erwarteten Häufigkeit des seltensten Zelltyps von Interesse ab.
Welche Sequenzierungstiefe ist für scRNA-seq erforderlich?
Für die Genanalyse sind 20.000-50.000 Reads pro Zelle Standard. Für die Isoform- oder Spleißanalyse können 50.000-100.000 Reads pro Zelle erforderlich sein. Eine höhere Tiefe ermöglicht eine empfindlichere Erkennung von schwach exprimierten Genen, jedoch zu höheren Kosten pro Zelle.
Sollte ich Seurat oder Scanpy für die scRNA-seq-Analyse verwenden?
Beide liefern vergleichbare Ergebnisse für Standard-Workflows. Seurat (R) bietet mehr integrierte Funktionen für Integration und Visualisierung. Scanpy (Python) bietet größere Flexibilität für benutzerdefinierte Analysen und ist speichereffizienter für Datensätze mit mehr als 50.000 Zellen.
Wie gehe ich mit Batch-Effekten in scRNA-seq-Daten um?
Harmony wird für die meisten Datensätze empfohlen. Die CCA-Integration von Seurat ist für Datensätze mit starken Batch-Effekten und überlappenden Zelltypen geeignet. MNN eignet sich für die Integration über verschiedene Plattformen oder Technologien hinweg.
Was ist der Unterschied zwischen 3' und 5' scRNA-seq?
3' scRNA-seq (Standard 10x Genomics) sequenziert das 3'-Ende von Transkripten zu den niedrigsten Kosten pro Zelle. 5' scRNA-seq sequenziert das 5'-Ende und ermöglicht die gekoppelte Profilierung von Immunrezeptoren neben der Genexpression, was es zur bevorzugten Wahl für immunologische Studien macht.
Kann ich scRNA-seq mit anderen Omics-Technologien kombinieren?
Ja. CITE-seq fügt die Quantifizierung von Oberflächenproteinen hinzu, scATAC-seq fügt die Chromatinzugänglichkeit hinzu, und die räumliche Transkriptomik bietet den Gewebe-Kontext. Die Integration von Multi-Omics ist ein aktives Forschungsfeld mit schnell verbesserten rechnerischen Methoden.
Wie bestimme ich die optimale Clusterauflösung für mein Datenset?
Testauflösungen von 0,2 bis 1,2 und bewerten Sie die Clusterqualität mithilfe des Silhouette-Scores, der differentiellen Expression zwischen Clustern und der biologischen Interpretierbarkeit von Marker-Genen. Die optimale Auflösung erzeugt Cluster, die transkriptionell unterschiedlich sind und bekannten Zelltypen entsprechen.
Was ist der Unterschied zwischen UMAP und t-SNE für die Visualisierung von scRNA-seq?
UMAP ist schneller, bewahrt die globale Struktur besser und ist der aktuelle Standard für die Visualisierung von scRNA-seq. t-SNE ist hervorragend darin, die lokale Struktur zu bewahren, kann jedoch die Beziehungen zwischen Clustern verzerren und ist bei großen Datensätzen langsamer.
Wie validiere ich Zelltypannotationen in scRNA-seq?
Verwenden Sie mehrere unabhängige Markergene für jeden Zelltyp, vergleichen Sie die automatisierte Annotation mit der manuellen Inspektion und validieren Sie diese anhand veröffentlichter Datensätze oder unabhängiger experimenteller Methoden.
Nur für Forschungszwecke, nicht zur klinischen Diagnose, Behandlung oder individuellen Gesundheitsbewertung gedacht.
Referenzen:
- Best Practices für die Einzelzellanalyse über Modalitäten hinweg. Nature Reviews Genetics2023;24:550-572.
- Aktuelle Best Practices in der Analyse von Einzelzell-RNA-Sequenzierungen: Ein Tutorial. Molekulare Systembiologie. 2019;15:e8746.
- Ein praktisches Handbuch zur Qualitätskontrolle von Einzelzell-RNA-Sequenzierungsdaten. Zeitschrift der Formosanischen Medizinischen Vereinigung. 2024;123:1205-1215.
- Fortschritte und Herausforderungen bei der Analyse von Einzelzell-RNA-Sequenzierungsdaten. Briefings in Bioinformatik2026;27:bbaf723.
- Einzelzell-Sequenzierung zu Multi-Omics: Technologien und Herausforderungen. Biomarkerforschung2024;12:124.


