
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Ein aktualisierter Leitfaden zur Illumina-Bibliotheksvorbereitung: Kits, Methoden und Strategien für herausfordernde Probenarten
Die Bibliotheksvorbereitung ist der wichtigste Faktor für den Erfolg der Sequenzierung. Unabhängig davon, wie leistungsstark die Sequenzierungsplattform oder wie ausgeklügelt die Datenanalyse-Pipeline ist, wird eine schlecht vorbereitete Bibliothek unbrauchbare Daten, verschwendete Flusszellkapazität und verlorene Zeit produzieren.
Aber im Jahr 2026 besteht die Herausforderung für die meisten Forscher nicht mehr darin, die grundlegenden Schritte der Bibliotheksvorbereitung zu verstehen. Die Herausforderung besteht darin, die richtige Methode aus einem zunehmend überfüllten Angebot auszuwählen – PCR-basiert vs. PCR-frei, fragmentierungsbasiert vs. tagmentierungsbasiert und Kits, die für spezifische Probenarten wie FFPE-Gewebe, zirkulierende zellfreie DNA (cfDNA) und Proben mit extrem niedrigem Input optimiert sind.
Dieser Leitfaden bietet einen praktischen Rahmen für die Entscheidungsfindung. Er behandelt die wichtigsten Bibliotheksvorbereitungsstrategien und -kits, die für die Illumina-Sequenzierung verfügbar sind, mit einem Schwerpunkt auf der Auswahl des richtigen Ansatzes für Ihren Proben-Typ, den Projektumfang und die Anforderungen an die Datenqualität.
Warum die Wahl der Bibliotheksvorbereitung wichtiger ist als je zuvor
Die Bibliotheksvorbereitung macht den Großteil der Variabilität in NGS-Workflows aus. Eine gut gestaltete Bibliothek produziert konsequent hochwertige Daten; eine schlecht gestaltete kann selbst auf dem besten Sequenziergerät versagen. Da die Sequenzierungskosten gesunken sind, hat die Bibliotheksvorbereitung einen verhältnismäßig größeren Anteil an den Gesamtkosten des Projekts eingenommen, was die Wahl der Methode zu einer bedeutenden finanziellen Entscheidung macht.
Drei Schlüsselfaktoren bestimmen die Wahl der Bibliotheksvorbereitungsmethode:
- Musterqualität und -mengeHochwertige genomische DNA (>1 µg) ermöglicht den Zugang zu PCR-freien Methoden, die Verzerrungen minimieren. Proben von niedriger Qualität oder geringer Menge (FFPE, cfDNA, Einzelzellen) erfordern spezialisierte Kits mit Reparaturenzymen oder Protokollen für ultra-niedrige Eingaben. Die häufigste Ursache für das Scheitern von Bibliotheken ist degradierendes Eingabematerial, das erst nach der Quantifizierung der Bibliothek identifiziert wurde – weshalb sich eine Investition in rigorose Qualitätskontrollen im Vorfeld vielfach auszahlt.
- ProjekttypDie Ganzgenomsequenzierung (WGS) erfordert eine geringe experimentelle Verzerrung und konsistente Abdeckung. Die gezielte Sequenzierung benötigt eine hohe Erfassungsrate. Die Amplicon-Sequenzierung basiert auf ausgewogenen Multiplex-Primern. Jeder Anwendungstyp stellt unterschiedliche Anforderungen an die Bibliotheksvorbereitungsmethode, was bedeutet, dass es kein einzelnes „bestes“ Kit gibt – nur das beste Kit für eine bestimmte Kombination aus Probe und Anwendung.
- Durchsatz und DurchlaufzeitEinige Methoden können in weniger als 3 Stunden von DNA zu einer sequenzierbereiten Bibliothek gelangen; andere benötigen 6–8 Stunden. Bei großen Chargen wird die Automatisierungsfähigkeit zu einem wichtigen Faktor. Ein Labor, das 96 Proben pro Woche verarbeitet, hat andere Durchsatzanforderungen als eines, das 16 Proben pro Monat verarbeitet.
Über diese Faktoren hinaus gewinnt ein viertes Kriterium an Bedeutung: die Komplexität der Bibliothek. Eine Bibliothek mit hoher Komplexität – das bedeutet, sie repräsentiert das ursprüngliche Genom oder Transkriptom mit minimaler Duplikation – erzeugt qualitativ hochwertigere Variantenaufrufe, zuverlässigere Expressionsmessungen und reproduzierbarere Ergebnisse über verschiedene Chargen hinweg. Bibliotheksvorbereitungsverfahren, die die Komplexität bewahren (PCR-frei, Niedrigzyklus-PCR und optimierte Tagmentierungsprotokolle), werden zunehmend für Projekte bevorzugt, bei denen die Datenqualität im Vordergrund steht.
Für Forscher, die ihr erstes NGS-Projekt planen oder eine bestehende Pipeline optimieren möchten, ist das Verständnis dieser Kompromisse entscheidend. Umfassend NGS-Dienste alle Methoden zur Bibliotheksvorbereitung abdecken, sodass es möglich ist, den optimalen Ansatz für die spezifischen Anforderungen jedes Projekts auszuwählen.
Der Standardarbeitsablauf in Kürze
Die grundlegenden Schritte der Illumina-Bibliotheksvorbereitung bleiben über die meisten Methoden hinweg konsistent, obwohl die spezifische Umsetzung variiert:
- FragmentierungDNA wird durch mechanisches Scheren, enzymatische Verdauung oder Tagmentation in Zielgrößenfragmente (typischerweise 200–800 bp) zerbrochen.
- Endreparatur und A-TailingFragmentenden sind abgestumpft, phosphoryliert und mit einem A-Schwanz versehen, um die Adapterligatur zu ermöglichen.
- Adapter-LigationSequenzierungsadapter, die P5/P7-Sequenzen, Index-Barcodes und Bindungsstellen für Sequenzierungsprimer enthalten, werden an die Fragmente ligiert.
- GrößenauswahlFragmente außerhalb des Zielgrößenbereichs werden entfernt, typischerweise unter Verwendung von SPRI-Magnetperlen.
- Bibliotheksamplifikation (optional)Die PCR-Amplifikation fügt genügend Material für die Sequenzierung hinzu. PCR-freie Methoden überspringen diesen Schritt vollständig.
- Bibliotheks-QCDie endgültige Bibliothek wird quantifiziert und qualitätsgeprüft mittels qPCR, fluorometrischen Assays und Kapillarelektrophorese (Bioanalyzer oder TapeStation).
Dieser Überblick ist absichtlich kurz gehalten, da die detaillierten Abläufe jedes Schrittes bereits in bestehenden Ressourcen behandelt werden. Der Fokus dieses Leitfadens liegt auf der Auswahl zwischen den verfügbaren Methoden und nicht auf dem Schritt-für-Schritt-Protokoll.
 Abbildung 1. Entscheidungsrahmen für die Bibliotheksvorbereitung – von Probenart und Projektzielen zur optimalen Vorbereitungsstrategie
Abbildung 1. Entscheidungsrahmen für die Bibliotheksvorbereitung – von Probenart und Projektzielen zur optimalen Vorbereitungsstrategie
Entscheidungsrahmen, der Forscher von Probenart, Eingabemenge und Projektzielen zur optimalen Bibliotheksvorbereitungsstrategie über PCR-basierte, PCR-freie und Tagmentationsmethoden leitet.
Die richtige Bibliotheksvorbereitungsstrategie wählen – PCR-basiert, PCR-frei oder Tagmentation
Jede Illumina-Bibliotheksvorbereitungsmethode fällt in eine von drei technischen Kategorien. Das Verständnis der Unterschiede zwischen diesen Kategorien ist der erste Schritt, um das richtige Kit für Ihr Projekt auszuwählen.
PCR-basierte Fragmentierung + LigationDNA wird durch mechanisches Scheren (Covaris) oder enzymatische Verdauung fragmentiert, gefolgt von Endreparatur, A-Tailing, Adapterligatur und PCR-Amplifikation. Dies ist der flexibelste und am weitesten verbreitete Ansatz. Er funktioniert gut über ein breites Spektrum an Eingabemengen (0,1 ng bis 1 µg) und Probenarten. Der Nachteil ist, dass die PCR-Amplifikation Verzerrungen in GC-reichen Regionen einführen und die Duplikationsraten erhöhen kann. Kits, die diese Strategie verwenden, benötigen typischerweise 4–8 Stunden für den vollständigen Workflow.
PCR-freie Fragmentierung + LigationDer gleiche Workflow wie oben, jedoch wird die PCR-Amplifikation weggelassen. Dies beseitigt PCR-eingeführte Verzerrungen und sorgt für die gleichmäßigste Genomabdeckung, was es zum Goldstandard für WGS-Anwendungen macht. Die Einschränkung besteht darin, dass PCR-freie Methoden eine höhere Eingangs-DNA erfordern (typischerweise >100 ng bis >1 µg, abhängig vom Kit), da es keinen Amplifikationsschritt gibt, um den Bibliotheksausstoß zu erhöhen.
Tagmentierung (transposase-basiert)Ein modifiziertes Transposase-Enzym fragmentiert gleichzeitig DNA und fügt Adaptersequenzen in einem einzigen Reaktionsschritt ein. Dies reduziert die Hands-on-Zeit und die Eingabebedürfnisse erheblich – einige Tagmentierungs-Kits arbeiten mit nur 1 ng Eingangs-DNA. Der Nachteil ist, dass Tagmentierungsmethoden sequenzabhängige Verzerrungen einführen können, insbesondere in Regionen mit niedrigem GC-Gehalt, und möglicherweise Bibliotheken mit einer engeren Insertgrößenverteilung erzeugen.
AuswahlrahmenBasierend auf der obigen Analyse kann ein praktischer Leitfaden zur Methodenauswahl wie folgt zusammengefasst werden:
- Hochwertige DNA >1 µg für WGS → PCR-freie Fragmentierung + Ligation produziert die gleichmäßigste Abdeckung mit den niedrigsten Duplizierungsraten
- Eingabe 10–100 ng, schnelle Bearbeitung erforderlich → Tagmentationsbasiert Methoden sind am effizientesten und reduzieren die Bibliotheksvorbereitungszeit um etwa die Hälfte.
- FFPE, cfDNA oder andere herausfordernde Proben → PCR-basierte Fragmentierung + Ligation mit Reparaturenzymen oder spezialisierten Ultra-Niedrig-Eingangskits
- Flexibles, vielseitiges Labor zur Verarbeitung verschiedener Probenarten PCR-basierte Fragmentierung + Ligation bietet den größten Eingangsbereich und die beste Anwendungskompatibilität
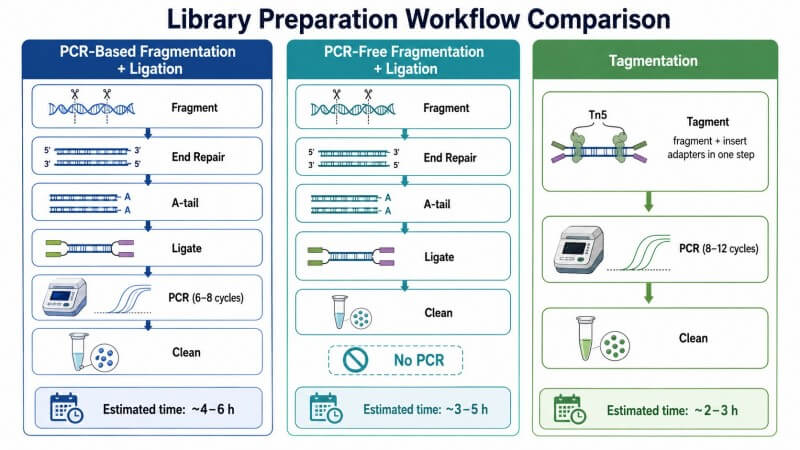 Abbildung 2. Drei Strategien zur Bibliotheksvorbereitung — PCR-basierte Fragmentierung + Ligation, PCR-frei und Tagmentation
Abbildung 2. Drei Strategien zur Bibliotheksvorbereitung — PCR-basierte Fragmentierung + Ligation, PCR-frei und Tagmentation
Überschrift: Vergleich der drei wichtigsten Illumina-Bibliotheksvorbereitungsstrategien, der Unterschiede im Fragmentierungsmechanismus, den Anforderungen an die Amplifikation, dem Eingangs-DNA-Bereich und den am besten geeigneten Anwendungen für jeden Ansatz.
Überlegungen zum LaborablaufNeben den oben genannten technischen Faktoren sollten auch praktische Laborlogistik die Wahl beeinflussen. PCR-basierte Fragmentierungs- und Ligationkits liefern konsistente Ergebnisse über eine Vielzahl von Probenarten hinweg, was sie ideal für zentrale Einrichtungen oder Dienstlabore macht, die unterschiedliche Probenarten erhalten. Tagmentationsmethoden sind besser für Labore geeignet, die eine konsistente Probenart in großem Maßstab verarbeiten und Geschwindigkeit priorisieren. PCR-freie Methoden sind am besten für Labore geeignet, die zuverlässigen Zugang zu hochwertiger, hochquantitativer DNA haben und einen spezifischen Bedarf an der Verzerrungsreduktion haben, die sie bieten.
Vergleich der verfügbaren Bibliotheksvorbereitungs-Kits nach wichtigen Parametern
Mehrere kommerziell erhältliche Kits setzen jede der drei oben genannten Strategien um. Die Wahl zwischen ihnen hängt von den spezifischen Anforderungen Ihres Projekts ab.
| Kit-Typ | Methode | Eingabebereich | Typische Zeit | Beste Anwendung |
|---|---|---|---|---|
| PCR-frei (Standardfragmentierung + Ligation) | Sonikation + Ligation, keine PCR | 100 ng – 1 µg | ~6 Std. | WGS, wo minimale Verzerrung entscheidend ist; hochqualitative DNA |
| PCR-basiert (Standardfragmentierung + Ligation) | Sonikation/enzymatisch + Ligation + PCR | 0,1 ng – 1 µg | ~4–6 Std. | Breiteste Anwendungsbereich; flexible Eingabe; FFPE mit Reparatur |
| Tagmentationsbasiert | Transposase-Fragmente + Adaptereinfügung | 1–50 ng | ~3 Std. | Niedriger Input; schneller Arbeitsablauf; Bakterien, Viren, kleine Genome |
| Ultra-niedrig-input / cfDNA-optimiert | Stem-Schleife oder spezialisierte Ligation | 0,05–50 ng | ~2,5–3 Std. | cfDNA, flüssige Biopsie, Einzelzelle, Sub-Nanogramm-Eingaben |
Praktische AuswahlberatungFür Labore, die eine Vielzahl von Probenarten verarbeiten, ist ein PCR-basiertes Fragmentierungs- und Ligation-Kit mit einem breiten Eingangsbereich die vielseitigste Wahl, die alles von hochwertigem gDNA bis hin zu FFPE, cfDNA und RNA-seq-Bibliotheken abdeckt. Für Labore, die sich ausschließlich auf hochwertige DNA-WGS konzentrieren, bietet ein PCR-freier Ansatz die beste Datenqualität mit den niedrigsten Duplikationsraten. Für Labore, die Geschwindigkeit und niedrige Eingangsmenge bei einfachen Probenarten priorisieren, ist Tagmentierung eine ausgezeichnete Option, die die praktische Arbeitszeit um bis zu 50 % reduzieren kann. Für Projekte, die cfDNA oder andere Anwendungen mit ultra-niedriger Eingangsmenge betreffen, sollte ein speziell für diese Eingaben optimiertes Kit die erste Überlegung sein – die Verwendung eines standardmäßigen PCR-basierten Kits bei cfDNA führt häufig zu Kontaminationen durch Adapter-Dimere, da die kurzen Insertfragmente nicht genügend Abstand zu den Produkten der Adapter-Adapter-Ligation bieten.
KostenüberlegungenDie Kosten pro Bibliothek variieren je nach den drei Strategien. PCR-basierte Fragmentierung + Ligation-Kits haben typischerweise die höchsten Kosten pro Reaktion, während Tagmentations-Kits oft günstiger sind. Die Kosten pro Bibliotheks-Kit sollten jedoch gegen die Kosten eines fehlgeschlagenen Sequenzierungslaufs abgewogen werden. Eine kleine Einsparung bei den Bibliotheksreagenzien ist unbedeutend, wenn sie eine Bibliothek produziert, die schlecht clusteriert, eine niedrige Datenausbeute liefert oder Verzerrungen einführt, die die biologische Interpretation beeinträchtigen. Die Gesamtkosten des Projekts – nicht die Kosten der Reagenzien pro Bibliothek – sind die relevante wirtschaftliche Kennzahl.
Sonderprobenart 1 — FFPE und degradierte DNA
FFPE-abgeleitetes DNA stellt einzigartige Herausforderungen bei der Bibliotheksvorbereitung dar. Die Formalinfixierung verursacht Quervernetzungen, die DNA fragmentieren und Basismodifikationen einführen – insbesondere die Cytosin-Deaminierung (C→T), die in Sequenzierungsdaten als scheinbare Mutationen erscheinen kann.
Erfolgreiche FFPE-Bibliotheksvorbereitung erfordert zwei spezifische Anpassungen:
- Schadenreparatur vor dem Bau der BibliothekEin Reparaturschritt vor der Bibliotheksvorbereitung unter Verwendung eines Enzymgemisches, das Uracil-DNA-Glykosylase (UDG) und andere Reparaturenzyme enthält, entfernt deaminierte Cytosine und repariert Nicks. Dieser Schritt ist entscheidend, um C→T-Artefakte zu beseitigen, die andernfalls als falsch-positive Varianten erscheinen würden.
- Eingabeflexibilität und reduzierte ZyklusanzahlFFPE-DNA ist typischerweise auf eine durchschnittliche Größe von 200–400 bp fragmentiert, was bereits im Zielbereich für die meisten Illumina-Bibliotheken liegt. Der Schwerpunkt sollte darauf liegen, zusätzliche Amplifikationszyklen zu minimieren, um weitere Verzerrungen zu vermeiden. PCR-basierte Fragmentierungs- und Ligation-Kits mit Reparaturmodulen sind der empfohlene Ansatz.
FFPE-Bibliotheken zeigen typischerweise eine breitere Insertgrößenverteilung und eine geringere Komplexität als Bibliotheken aus hochqualitativem DNA. Ein Bioanalyzer-Trace, der einen breiten Peak von 150–600 bp zeigt, anstatt eines scharfen Peaks, ist für FFPE-Proben normal. Das wichtigste Qualitätskontrollmaß ist nicht die Peakform, sondern das Vorhandensein eines klaren Bibliothekspeaks über dem Signal von Adapter-Dimeren.
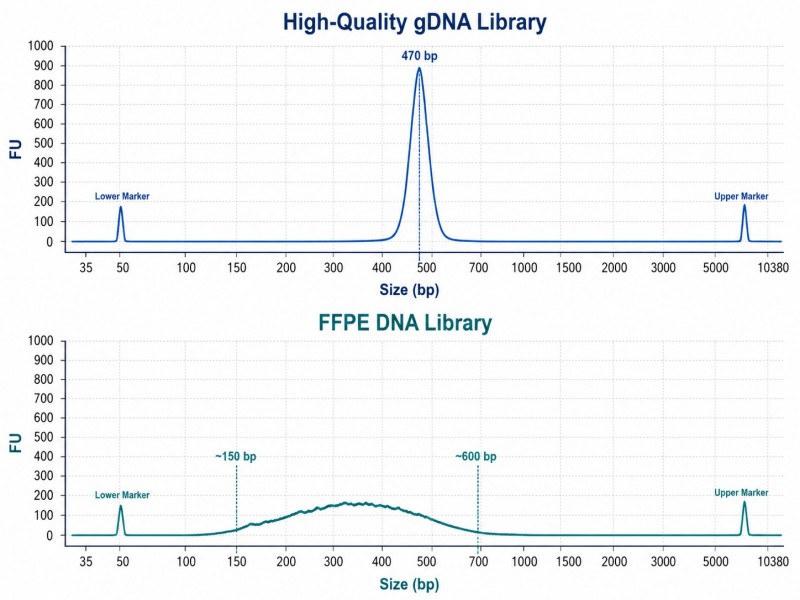 Abbildung 3. Vergleich der FFPE-DNA-Bioanalyzer-Spuren — degradierte FFPE-DNA im Vergleich zu hochqualitativen genomischen DNA-Bibliotheksspuren
Abbildung 3. Vergleich der FFPE-DNA-Bioanalyzer-Spuren — degradierte FFPE-DNA im Vergleich zu hochqualitativen genomischen DNA-Bibliotheksspuren
Bildunterschrift: Vergleichendes Bioanalyzer-Elektrophoregramm, das die breite Peakform zeigt, die typisch für aus FFPE gewonnenen DNA-Bibliotheken ist, im Gegensatz zu dem scharfen, gut definierten Peak von hochqualitativen genomischen DNA-Bibliotheken.
Sonderproben Typ 2 — cfDNA und Ultra-Niedrig-Eingabeproben
Zirkulierendes zellfreies DNA (cfDNA) ist in Plasma in sehr niedrigen Konzentrationen vorhanden (typischerweise 1–50 ng pro mL Blut) und besteht aus Fragmenten mit einer durchschnittlichen Länge von etwa 167 bp – der Länge von DNA, die um ein einzelnes Nukleosom gewickelt ist. Diese Eigenschaften erfordern einen grundlegend anderen Ansatz zur Bibliotheksvorbereitung als bei genomischer DNA.
Wichtige Überlegungen zur Vorbereitung von cfDNA-Bibliotheken:
- Adapter-Dimer-SteuerungDa cfDNA-Fragmente kurz sind (~167 bp + Adapter = ~300 bp endgültige Bibliothek), liegt jede Kontamination durch Adapter-Dimere (~120 bp) im gleichen Größenbereich und kann nicht effektiv durch Größenauswahl entfernt werden. Kits mit Stem-Loop- oder Bubble-Adapter reduzieren die Dimersbildung effektiver als Standard-Y-Adapter für cfDNA-Anwendungen.
- PCR-ZyklusoptimierungDer cfDNA-Eingang ist für PCR-freie Methoden zu niedrig, daher ist eine Amplifikation erforderlich. Allerdings birgt jeder zusätzliche PCR-Zyklus das Risiko, Verzerrungen und Duplikate einzuführen. Das optimale Gleichgewicht liegt typischerweise bei 8–12 Zyklen, kalibriert, um einen ausreichenden Bibliotheksausstoß zu erzeugen, ohne die Duplikationsraten von 10–15 % zu überschreiten.
- Index-Hopping-EmpfindlichkeitDa jedes Molekül in einer cfDNA-Bibliothek wertvoll ist, ist Index-Hopping, das Reads zwischen Proben falsch zuordnet, besonders schädlich. Einzigartige doppelte Indizes (UDI) werden für multiplexe cfDNA-Projekte dringend empfohlen.
Das cfDNA-Bibliotheksprofil auf einem Bioanalyzer sollte einen Peak bei etwa 280–330 bp (167 bp Insert + 120–140 bp Adapter) zeigen. Ein zweiter Peak bei 120–140 bp weist auf eine Kontamination mit Adapter-Dimeren hin und deutet auf die Notwendigkeit einer Protokolloptimierung hin.
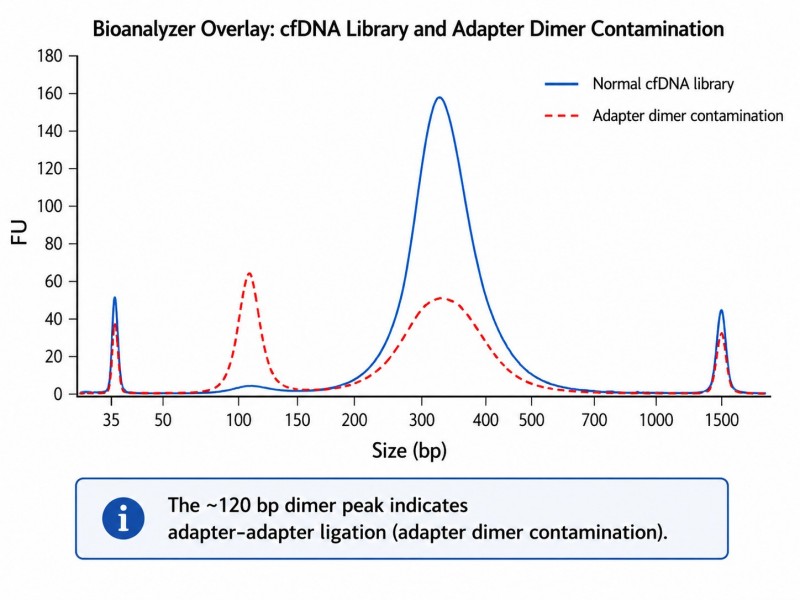 Abbildung 4. cfDNA-Bibliotheks-Bioanalyzer-Trace — typischer Bibliothekspeak und Adapter-Dimer-Kontamination
Abbildung 4. cfDNA-Bibliotheks-Bioanalyzer-Trace — typischer Bibliothekspeak und Adapter-Dimer-Kontamination
Beschriftung: Bioanalysator-Spur einer cfDNA-Bibliothek, die den erwarteten Bibliothekspeak bei ~280–330 bp und einen kleineren Adapter-Dimer-Peak bei ~120–140 bp zeigt, der auf eine Kontamination hinweist, die eine Optimierung des Protokolls erfordert.
Vergleich: cfDNA vs. standardisierte gDNA-BibliotheksvorbereitungDie grundlegenden Unterschiede zwischen der Bibliotheksvorbereitung von cfDNA und gDNA gehen über die Eingangsmenge hinaus. Da cfDNA-Fragmente bereits kurz sind und charakteristische Enden aufweisen (5-Phosphat, 3-Hydroxylgruppen aus der nucleosomalen Spaltung), wird der Fragmentierungsschritt vollständig weggelassen. Der Workflow zur Bibliotheksvorbereitung beginnt direkt mit der Endreparatur und dem A-Tailing, gefolgt von der Adapterligatur. Auch die Aufräumstrategie unterscheidet sich: Die Verhältnisse der SPRI-Perlen müssen sorgfältig optimiert werden, um die kurzen cfDNA-Fragmente zu behalten und gleichzeitig Adapter-Dimere zu entfernen, die nur etwa 40–60 bp kürzer sind als die Zielbibliotheksfragmente. Eine doppelseitige Perlenaufreinigung – zuerst bei 0,6–0,8×, um große Fragmente zu entfernen, dann bei 1,5–1,8×, um die cfDNA-Bereichsfragmente zu erfassen – ist ein gängiger und effektiver Ansatz.
WGS vs. gezielte Anreicherung vs. Amplicon — Wie sich die Bibliotheksvorbereitung je nach Anwendung unterscheidet
Der Workflow zur Bibliotheksvorbereitung ändert sich je nachdem, was danach kommt.
| Anwendung | Vorbereitungsziel | Wichtiger QC-Metrik | Eingabebedarf |
|---|---|---|---|
| Whole-Genome-Sequenzierung | Einheitliche Abdeckung mit minimaler Verzerrung | Einfügen der Größenverteilung, Duplikationsrate | 100 ng – 1 µg; PCR-frei bevorzugt |
| Gezielte Anreicherung (WES, Panels) | Effiziente Erfassung von Zielregionen | % Zielgenauigkeit, Abdeckungsuniformität | 10–200 ng |
| Amplicon-Sequenzierung | Ausgewogene Verstärkung über Ziele hinweg | Lesen des Gleichgewichts über Amplicons, Primer-Dimer | 1–100 ng |
Für WGS-BibliotheksvorbereitungDie Priorität liegt darin, experimentelle Verzerrungen zu minimieren. PCR-freie Methoden werden bevorzugt, wenn die Eingangsmenge dies zulässt. Die kritischen Qualitätskontrollpunkte sind die Fragmentgrößenverteilung (ein enger Peak bei der Zielgröße) und die Duplikationsrate (sollte für PCR-freie Methoden <10 % und für PCR-basierte Methoden <15 % betragen).
Für gezielte Anreicherung BibliotheksvorbereitungDie ursprüngliche Bibliothek wird auf die gleiche Weise wie eine WGS-Bibliothek erstellt, jedoch wird ein zusätzlicher Hybridisierungsfangschritt hinzugefügt, um Zielregionen zu erfassen. Die entscheidende Qualitätskontrollmetrik wechselt von der Gleichmäßigkeit der Abdeckung zur Fangeffizienz (% der Reads, die auf Zielregionen abgebildet werden). Die Bibliotheksvorbereitung für gezielte Anreicherung muss genügend Komplexität erzeugen, um zu vermeiden, dass doppelte Reads die On-Target-Daten nach dem Fang dominieren.
Für Amplicon-basierte BibliotheksvorbereitungDie Bibliothek wird durch Multiplex-PCR anstelle von Fragmentierung und Adapterligatur erzeugt. Die Hauptschwierigkeit besteht darin, die Primerleistung über alle Ziele hinweg auszubalancieren, um das Auslassen spezifischer Regionen zu vermeiden. Die Qualitätskontrolle konzentriert sich auf die Gleichmäßigkeit der Abdeckung über das Zielset – eine Standardmetrik ist der Prozentsatz der Amplicons, die innerhalb von 0,2×–2× der durchschnittlichen Abdeckungstiefe liegen.
Eine Anmerkung zur RNA-BibliotheksvorbereitungWährend der Schwerpunkt dieses Leitfadens auf der DNA-Bibliotheksvorbereitung liegt, folgt die RNA-seq-Bibliotheksvorbereitung einem anderen Workflow. Anstelle der Fragmentierung vor der Adapterligierung wird RNA zunächst durch reverse Transkription in cDNA umgewandelt und dann fragmentiert (oder vor der reversen Transkription fragmentiert, je nach Protokoll). Die Schritte der Adapterligierung und Amplifikation sind ähnlich wie bei der DNA-Bibliotheksvorbereitung, aber die Strangspezifität muss beibehalten werden, wenn das experimentelle Design erfordert, den ursprünglichen RNA-Strang von seinem Komplement zu unterscheiden.
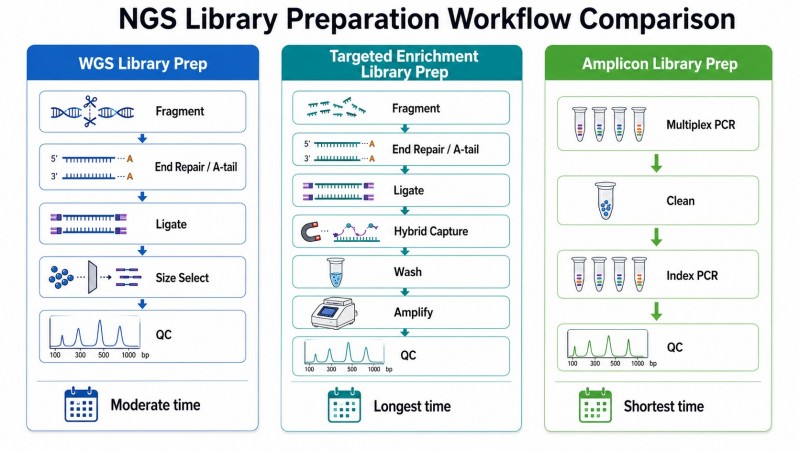 Abbildung 5. WGS-Bibliotheksvorbereitung vs. gezielte Anreicherungsbibliotheksvorbereitung vs. Amplicon-Bibliotheksvorbereitung — Workflow-Vergleich
Abbildung 5. WGS-Bibliotheksvorbereitung vs. gezielte Anreicherungsbibliotheksvorbereitung vs. Amplicon-Bibliotheksvorbereitung — Workflow-Vergleich
Bildunterschrift: Vergleichendes Workflow-Diagramm, das die drei Hauptwege zur NGS-Bibliotheksvorbereitung zeigt – WGS, gezielte Anreicherung und Amplicon – mit Unterschieden in der Fragmentierungsstrategie, den Erfassungs-/Amplifikationsschritten und den wichtigsten QC-Metriken für jeden.
Mehrere kommerziell erhältliche RNA-Bibliotheksvorbereitungskits erreichen dies durch unterschiedliche chemische Strategien. Die Qualitätskontrolle von RNA-seq-Bibliotheken muss auch die Effizienz der ribosomalen RNA-Depletion bewerten, die ein kritisches Qualitätsmerkmal ist, das für die DNA-Bibliotheksvorbereitung nicht zutrifft.
Multiplexing-Strategie für Batch-ProjekteBei der Vorbereitung von Bibliotheken für ein Multi-Proben-Projekt muss die Barcode-Strategie geplant werden, bevor mit dem Bau der Bibliothek begonnen wird. Für Projekte mit 96 oder weniger Proben pro Sequenzierlauf ist in der Regel eine einfache Indizierung ausreichend, vorausgesetzt, die Indexsequenzen weisen eine angemessene Diversität auf. Für Projekte mit mehr als 96 Proben oder wenn Multiplex-Läufe zur Analyse kombiniert werden, werden eindeutige doppelte Indizes (UDI) dringend empfohlen. UDI beseitigt das Risiko des Index-Hoppings, das zu falsch-positiven Variantenaufrufen in gepoolten Sequenzierungsdesigns führen kann. Die Wahl der Indizierungsstrategie beeinflusst den Workflow der Bibliotheksvorbereitung, da einige Indexsets spezifische Adapterkonfigurationen erfordern, die wiederum die Ligationchemie und das Reinigungsprotokoll beeinflussen.
Lesebibliothek QC-Ergebnisse — Ein praktischer Leitfaden zu häufigen Fehlermustern
Das Erlernen der Interpretation von Bioanalyzer- oder TapeStation-Spuren ist eine der nützlichsten Fähigkeiten für jeden, der mit NGS-Bibliotheken arbeitet. Hier sind die sechs häufigsten Spur-Muster und was sie bedeuten:
- Normale BibliothekEin einzelner, symmetrischer Gipfel im erwarteten Größenbereich. Für eine Standardbibliothek mit 350 bp Insert und Adaptern bedeutet dies einen Gipfel bei etwa 470 bp. Geringe Schultern auf beiden Seiten sind akzeptabel.
- Adapter-Dimer-SpitzeEin zweiter, kleinerer Peak bei 120–140 bp (abhängig vom Adapterdesign). Dies weist darauf hin, dass Adaptermoleküle während des Ligation-Schrittes miteinander ligiert wurden, anstatt an die Insert-DNA. Ein Dimer-Peak von <5% der gesamten Bibliotheksmasse ist allgemein akzeptabel; höhere Werte erfordern zusätzliche Reinigung oder eine Überarbeitung des Protokolls.
- Linksverschobener Gipfel (kurze Fragmente)Der Bibliothekspeak liegt unter dem erwarteten Größenbereich. Dies deutet normalerweise auf eine Überfragmentierung während des Scher- oder Tagmentationsschrittes hin. Es verringert die kartierbare Lese-Länge und kann eine Re-Optimierung der Fragmentierungsbedingungen erforderlich machen.
- Rechtsverschobener Gipfel (lange Fragmente)Der Bibliothekspeak liegt über dem erwarteten Größenbereich. Dies weist auf eine Unterfragmentierung oder eine ineffiziente Größenwahl hin. Diese Bibliotheken können eine niedrigere Clusterdichte auf dem Flusszelle erzeugen, da längere Fragmente nicht so effizient amplifiziert werden.
- Breiter Gipfel (breite Größenverteilung)Die Spur erstreckt sich über mehr als 500 bp ohne dominanten Peak. Dies ist häufig bei degradiertem Eingangs-DNA, insbesondere bei FFPE, der Fall. Es ist akzeptabel für FFPE-Bibliotheken, solange die Mehrheit der Fragmente innerhalb oder nahe der Sequenzierleselänge liegt.
- Kein Peak oder sehr schwaches SignalDie Bibliothek hat eine extrem niedrige Ausbeute oder ist vollständig fehlgeschlagen. Häufige Ursachen sind fehlgeschlagene Adapterligationen, unzureichendes Eingangs-DNA oder degradierte Ligationenzyme. Dies erfordert eine Wiederholung der Bibliotheksvorbereitung mit einer positiven Kontrolle, um die Ursache zu isolieren.
Beispiele für jedes Spur-Muster sind unten aufgeführt.
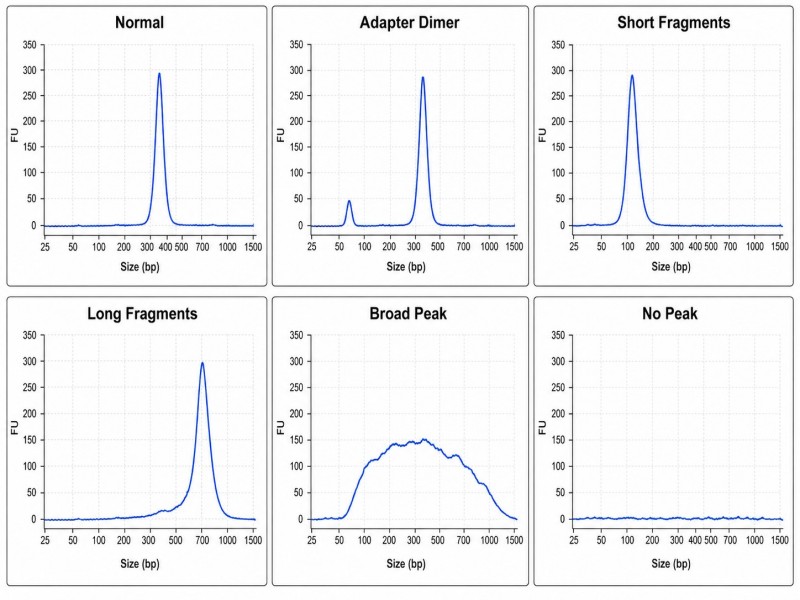 Abbildung 6. Normale vs. abnormale Bioanalyzer-Bibliotheksverläufe — sechs häufige Verlaufsmuster mit Anmerkungen
Abbildung 6. Normale vs. abnormale Bioanalyzer-Bibliotheksverläufe — sechs häufige Verlaufsmuster mit Anmerkungen
Bildunterschrift: Sechs häufige Bioanalyzer-Spurenmuster für NGS-Bibliotheken: normales Bibliotheksmaximum, Adapter-Dimer-Kontamination, linksverschobenes (überfragmentiertes), rechtsverschobenes (unterfragmentiertes), breites Maximum (degradierte Eingabe) und fehlgeschlagene Bibliothek ohne nachweisbares Signal.
Wie man die Ausbeute von Bibliotheken für Proben mit niedrigem Input optimiert
Eine der häufigsten praktischen Fragen, mit denen Forscher konfrontiert sind, ist, wie man den Bibliotheksausbeute maximieren kann, wenn das Ausgangsmaterial begrenzt ist. Die Standardempfehlung "verwenden Sie mehr Eingangs-DNA" ist oft keine Option, daher sind alternative Strategien erforderlich.
Verluste bei jedem Reinigungsschritt minimierenJeder SPRI-Perlen-Reinigungsschritt verliert 10–20 % des Bibliotheksmaterials, selbst unter optimalen Bedingungen. Bei Proben mit niedrigem Input kann die Reduzierung der Anzahl der Reinigungsschritte den endgültigen Ertrag erheblich verbessern. Einige Protokolle kombinieren die Reinigung nach der Ligation und nach der PCR in einen einzigen Perlschritt, was jedoch zu einer etwas breiteren Größenverteilung führt. Wenn der Input kritisch begrenzt ist (unter 10 ng), ist dieser Kompromiss in der Regel akzeptabel.
Optimierung der PCR-ZyklusanzahlDie Beziehung zwischen PCR-Zyklen und der Bibliotheksausbeute ist nicht linear. Die ersten paar Zyklen führen zu einer exponentiellen Amplifikation; nach 10–12 Zyklen bringen zusätzliche Zyklen relativ wenig Ausbeute, während sie die Duplikationsraten erheblich erhöhen. Bei sehr niedrigen Eingaben (<5 ng) können 12–14 Zyklen erforderlich sein. Bei Eingaben über 10 ng sind typischerweise 8–10 Zyklen ausreichend. Die Überwachung der Amplifikationskurve durch qPCR oder das Durchführen eines kleinen Pilotversuchs bei 8, 10, 12 und 14 Zyklen für einen neuen Proben-Typ hilft, das optimale Gleichgewicht zu identifizieren.
Verwenden Sie Trägersubstanzen.Das Hinzufügen eines Trägers (z. B. lineares Polyacrylamid oder Glykogen) während der Ethanolfällung kann die Rückgewinnung von DNA in niedrigen Konzentrationen verbessern. Dies ist besonders nützlich für cfDNA-Proben, bei denen die Eingabemengen im Bereich von 1–10 ng liegen und jedes Nanogramm zählt.
Betrachten Sie Einzelrohr-Workflows.Einige kürzlich entwickelte Methoden zur Bibliotheksvorbereitung führen Fragmentierung, Endreparatur, A-Tailing und Adapterligierung in einem einzigen Reaktionsgefäß ohne zwischengeschaltete Reinigungsschritte durch. Diese Einrohr-Workflows können die Umwandlungseffizienz für Proben mit niedrigem Input um 30–50 % im Vergleich zu mehrstufigen Protokollen verbessern, jedoch auf Kosten einer weniger präzisen Größenkontrolle.
Automatisierung und Skalierbarkeit in der Bibliotheksvorbereitung
Mit dem Wachstum von Sequenzierungsprojekten wird die manuelle Bibliotheksvorbereitung zu einem Engpass. Ein Labor, das 384 Bibliotheken pro Woche verarbeitet, kann es sich nicht leisten, jede Bibliothek von Hand vorzubereiten, und Batch-Effekte von verschiedenen Bedienern können unerwünschte technische Variationen einführen.
Flüssigkeitshandhabungs-Automatisierungsplattformen (wie die von Beckman Coulter, Hamilton oder Agilent) können 96 oder 384 Proben parallel mit konsistenten Reagenzvolumina und Inkubationszeiten verarbeiten. Die wichtigsten Überlegungen zur Einführung automatisierter Bibliotheksvorbereitung umfassen:
- ProtokollkompatibilitätNicht alle kommerziellen Bibliotheksvorbereitungs-Kits verfügen über validierte Automatisierungsprotokolle. Die Auswahl eines Kits mit einem etablierten Automatisierungsskript reduziert die Entwicklungszeit erheblich.
- VolumenbeschränkungenAutomatisierte Flüssigkeitshandhabungsgeräte haben minimale Pipettiervolumina (typischerweise 0,5–2 µL). Kits mit kleineren Reaktionsvolumina erfordern möglicherweise den Wechsel zu Versionen mit höherem Volumen für die Kompatibilität mit der Automatisierung.
- Handhabung von magnetischen PerlenDie automatisierte Perlen-basierte Reinigung ist der herausforderndste Schritt zur Optimierung. Die Sedimentationszeit der Perlen, die Magnetaktivierung und die Geschwindigkeit der Überstandentfernung beeinflussen alle die Reproduzierbarkeit.
- QC-IntegrationDie effizientesten automatisierten Workflows beinhalten Inline-QC-Schritte, wie automatisiertes Plattenlesen zur Quantifizierung, um fehlerhafte Bibliotheken zu identifizieren, bevor sie in die Sequenzierung übergehen.
Für Forschungsteams, die großangelegte Sequenzierungsprojekte aufbauen, NGS-Dienste mit automatisierten Bibliotheksvorbereitungsfähigkeiten kann die Durchsatzmenge und Reproduzierbarkeit bieten, die manuelle Methoden nicht erreichen können.
Ein praktischer QC-Workflow vor der Sequenzierung
Bevor eine Charge von Bibliotheken in einen Sequenzierungslauf überführt wird, sollte ein standardisierter QC-Workflow auf jede Bibliothek angewendet werden. Das Ziel ist es, Bibliotheken zu identifizieren, die wahrscheinlich fehlschlagen, bevor sie die Kapazität des Flusszellen verschwenden.
- Quantifizierung durch qPCRDies ist die genaueste Methode zur Bestimmung der Konzentration amplifizierbarer Bibliotheksmoleküle. Ein qPCR-Ergebnis, das sich mehr als um das Dreifache von der Qubit-Messung unterscheidet, weist häufig auf eine Kontamination mit Adapter-Dimeren oder auf fehlgeschlagene Ligation hin.
- Größenverteilung durch KapillarelektrophoreseEin Bioanalyzer- oder TapeStation-Trace bestätigt, dass die Fragmente im erwarteten Größenbereich liegen und dass die Adapterdimer-Level akzeptabel sind.
- MolaritätsberechnungDie Konzentration aus der qPCR wird in Molarität umgerechnet, indem die durchschnittliche Fragmentgröße aus dem Bioanalyzer-Tracé verwendet wird, die zur Berechnung des Ladevolumens dient.
- Pool-NormalisierungFür multiplexierte Läufe werden einzelne Bibliotheken in äquimolaren Mengen gemischt, wobei 10-20% Überschuss vorbereitet werden.
- Pilot-TitrationFür neuartige Bibliothekstypen wird dringend empfohlen, einen Pilotversuch mit 2-3 Ladekonzentrationen für den vorgesehenen Flusszelltyp durchzuführen.
Dieser QC-Workflow dauert etwa 2-3 Stunden für 96 Bibliotheken und sollte als routinemäßiger Bestandteil jedes NGS-Projekts betrachtet werden.
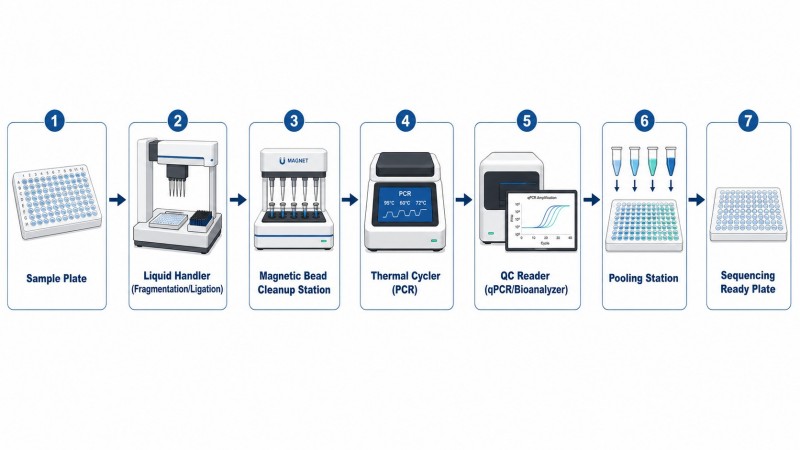 Abbildung 7. Automatisierter Arbeitsablauf für die Hochdurchsatz-Bibliotheksvorbereitung
Abbildung 7. Automatisierter Arbeitsablauf für die Hochdurchsatz-Bibliotheksvorbereitung
Automatisierter Flüssigkeitshandhabungs-Workflow für die Bibliotheksvorbereitung, der die wichtigsten Integrationspunkte zeigt: Protokollkompatibilität, Volumenbeschränkungen, Optimierung der Handhabung von magnetischen Perlen und Inline-QC für skalierbare Produktion.
Wie CD Genomics die Bibliotheksvorbereitung unterstützt
CD Genomics bietet umfassende Bibliotheksvorbereitungsdienste an, die das gesamte Spektrum der Illumina-kompatiblen Methoden und Probenarten abdecken.
Verfügbare MethodenUnser Labor führt PCR-basierte Fragmentierung + Ligation, PCR-freie und tagmentationsbasierte Bibliotheksvorbereitungsverfahren durch, unter Verwendung der am weitesten verbreiteten kommerziellen Kits. Die Wahl der Methode wird durch Ihren Proben-Typ und die Projektanforderungen bestimmt.
Spezielle ProbenexpertiseWir haben umfangreiche Erfahrungen mit der Reparatur von FFPE-DNA und der Bibliothekskonstruktion, der Vorbereitung von cfDNA und ultra-niedrigem Input sowie automatisierungsoptimierten Arbeitsabläufen für großangelegte Projekte gesammelt. Unsere Protokolle sind für Blut, Gewebe, FFPE, cfDNA, Einzelzellen sowie Pflanzen- und Mikrobiologiemuster validiert.
Bibliotheks-QCJede Bibliothek durchläuft eine strenge Qualitätskontrolle, einschließlich Bioanalyzer- oder TapeStation-Spuranalyse, qPCR-Quantifizierung und Bestätigung der Größenverteilung, bevor sie mit der Sequenzierung fortfährt.
SkalierbarkeitFür große Batch-Projekte setzen wir automatisierte Flüssigkeitshandhabungs-Workflows ein, die eine konsistente Bibliotheksqualität über alle Proben hinweg gewährleisten. Dies ist besonders wertvoll für Mehrbatch-Projekte, bei denen die Reproduzierbarkeit von Batch zu Batch entscheidend ist.
Für weitere Details erkunden Sie unser NGS-Dienste oder kontaktieren Sie unser Team für projektspezifische Empfehlungen.
Häufig gestellte Fragen
Was ist der Unterschied zwischen PCR-basierter und PCR-freier Bibliotheksvorbereitung?
PCR-basierte Bibliotheken umfassen einen Amplifikationsschritt, der den Ertrag erhöht, aber Verzerrungen in GC-reichen Regionen einführen und die Duplikationsraten erhöhen kann. PCR-freie Bibliotheken verzichten auf die Amplifikation, was eine gleichmäßigere Abdeckung zur Folge hat, erfordern jedoch eine höhere Eingangs-DNA (>100 ng).
Welche Bibliotheksvorbereitungsmethode ist am besten für FFPE-DNA-Proben?
Die auf PCR basierende Fragmentierung und Ligation mit einem vorhergehenden DNA-Reparaturschritt für Bibliotheken wird für FFPE-Proben empfohlen. Der Reparaturschritt entfernt Cytosin-Deaminierungsartefakte, die andernfalls als falsch-positive Mutationen erscheinen würden.
Kann die PCR-freie Bibliotheksvorbereitung mit DNA mit niedrigem Input verwendet werden?
Typischerweise nicht. PCR-freie Methoden erfordern 100 ng bis 1 µg Eingangs-DNA. Für Eingaben unter 100 ng ist eine PCR-basierte Methode erforderlich, um einen ausreichenden Bibliotheksausstoß zu erzeugen.
Wie sieht ein Adapter-Dimer in einem Bioanalyzer-Tracing aus?
Ein Adapterdimer erscheint als kleiner Peak bei etwa 120–140 bp, deutlich unter dem erwarteten Bibliothekspeak. Wenn der Dimer mehr als 5 % der gesamten Bibliotheksmasse ausmacht, wird eine zusätzliche Reinigung empfohlen.
Wie wähle ich zwischen Tagmentierung und Fragmentierung + Ligation?
Tagmentation bietet einen schnelleren Arbeitsablauf und geringere Eingabebedürfnisse, kann jedoch GC-Bias einführen. Fragmentierung + Ligation sorgt für eine gleichmäßigere Abdeckung über den GC-Gehalt und ist die bevorzugte Wahl für WGS und Anwendungen, die eine einheitliche genomische Repräsentation erfordern.
Was ist die minimale Eingangs-DNA für die standardmäßige Bibliotheksvorbereitung?
Standard-PCR-basierte Kits benötigen typischerweise 0,1–1 ng Mindestinput. Ultra-niedrig-input Kits können mit so wenig wie 50 pg arbeiten. PCR-freie Kits erfordern 100 ng–1 µg.
Wie entferne ich Adapter-Dimeren aus meiner Bibliothek?
Adapter-Dimere können reduziert werden, indem das Verhältnis der SPRI-Perlenreinigung optimiert wird (höhere Verhältnisse behalten mehr Dimere), eine doppelseitige Größenselektion verwendet wird oder ein gelbasiertes Größenselektionselement für hartnäckige Fälle hinzugefügt wird.
Welche QC-Metriken sollte ich für die WGS-Bibliotheksvorbereitung berichten?
Mindestens: Bibliothekskonzentration (aus qPCR), Fragmentgrößenverteilung (aus Bioanalyzer) und Duplikationsrate (aus einem QC-Lauf mit flacher Sequenzierung oder computergestützter Schätzung).
Kann CD Genomics Bibliotheken aus cfDNA vorbereiten?
Ja. Wir bieten cfDNA-optimierte Bibliotheksvorbereitung mit ultra-niedrigem Input-Protokoll und Stem-Loop-Adapter-Designs an, die die Bildung von Adapter-Dimeren in Kurzfragmentbibliotheken minimieren.
Wie lange dauert eine typische Bibliotheksvorbereitung?
Ein standardmäßiges PCR-basiertes Fragmentierungs- und Ligationprotokoll benötigt 4-6 Stunden von der DNA-Eingabe bis zur bereit zum Sequenzieren Bibliothek. Tagmentationsmethoden können in etwa 3 Stunden abgeschlossen werden. PCR-freie Methoden erfordern 5-7 Stunden aufgrund zusätzlicher Reinigungsschritte, die erforderlich sind, um nicht inkorporierte Adapter zu entfernen, ohne die Möglichkeit einer PCR-basierten Reinigung.
Was ist der Unterschied zwischen der Einzel-Index- und der Doppel-Index-Bibliotheksvorbereitung?
Einzelindexbibliotheken verwenden eine Barcode-Sequenz pro Probe, während Dual-Index-Bibliotheken zwei unabhängige Barcode-Sequenzen verwenden (eine auf jedem Adapter). Dual-Indexierung verringert das Risiko von Probenfehlzuweisungen, wenn mehrere Bibliotheken für das Sequenzieren zusammengelegt werden, insbesondere auf gemusterten Flusszellenplattformen, wo Index-Hopping häufiger vorkommt.
Was verursacht hohe Duplikationsraten in Sequenzierungsdaten?
Hohe Duplikationsraten (>20%) werden am häufigsten durch unzureichendes Eingangs-DNA, übermäßige PCR-Zyklen oder geringe Ausgangs-Bibliothekskomplexität verursacht. Bei WGS-Bibliotheken erzeugen PCR-freie Methoden die niedrigsten Duplikationsraten, während PCR-basierte Methoden mit Eingängen unterhalb des empfohlenen Bereichs am anfälligsten für dieses Problem sind.
Nur für Forschungszwecke.
Referenzen:
- Beste Praktiken für die Illumina-Bibliotheksvorbereitung. Aktuelle Protokolle in der Humangenetik2019;102(1):e86.
- Vergleich der Sequenzierungsverzerrung der derzeit verfügbaren Bibliotheksvorbereitungskits. DNA-Forschung2019;26(5):391-402.
- Vergleichende Analyse von Bibliotheksvorbereitungsansätzen für FFPE-DNA. Wissenschaftliche Berichte2025;15:12992.
- Eine vergleichende Analyse von Bibliotheksvorbereitungsansätzen für Proben mit niedrigem Input. BMC Genomik2018;19:763.


