
 Richtlinien zur Einreichung von Proben
Richtlinien zur Einreichung von Proben
Analyse von Kopienzahlvariationen (CNV): Nachweismethoden, Tiefenstrategien und bioinformatische Werkzeuge
Die Kopienzahlvariation (CNV) bezieht sich auf die Duplikation oder Deletion von DNA-Segmenten, die größer als 1 kb sind. Im menschlichen Genom machen CNVs mehr Gesamtdifferenzen in Basenpaaren zwischen Individuen aus als Einzel-Nukleotid-Varianten, doch sie sind schwieriger zu erkennen, da das primäre Signal – die Lesetiefe – eine kontinuierliche Variable ist, die von zahlreichen technischen Störfaktoren beeinflusst wird. Im Gegensatz zur SNV-Identifizierung, die von der Basenidentität an einer einzelnen Position abhängt, erfordert die CNV-Erkennung die Integration von Tiefeninformationen über genomische Fenster, die Korrektur systematischer Verzerrungen und die Segmentierung des Genoms in Regionen mit konsistenter Kopienzahl.
Dieser Leitfaden bietet einen praktischen Rahmen für Forscher, die Erfahrung mit NGS-Daten haben und CNV-Analyseprojekte entwerfen, durchführen und interpretieren müssen. Er behandelt die algorithmischen Grundlagen der Read-Depth-CNV-Erkennung, die quantitative Beziehung zwischen Sequenzierungstiefe und Nachweisempfindlichkeit, einen detaillierten Vergleich von Bioinformatik-Tools mit Benchmark-Leistungsdaten, die wichtigsten technischen Herausforderungen, die die Genauigkeit beeinträchtigen, und praktische Strategien für das Projektdesign – von Low-Pass-Screening bis hin zu hochauflösenden genomweiten Profilierungen. Der Schwerpunkt liegt darauf, wie analytische Entscheidungen die Arten und Größen von CNVs beeinflussen, die zuverlässig nachgewiesen werden können, und darauf, die häufigen Fallstricke zu vermeiden, die zu falsch-positiven oder falsch-negativen Ergebnissen führen. Ob Sie nun germline CNVs in einer Bevölkerungsstudie mit über 1.000 Proben oder somatische CNVs in einer kleinen Krebskohorte analysieren, die Prinzipien der GC-Normalisierung, Mappability-Filterung und kohortenbewussten Segmentierung gelten für alle Skalen der CNV-Analyse.
Whole-Genome-Sequenzierungsdienste Unterstützen Sie die CNV-Erkennung über alle Tiefenkonfigurationen hinweg – von hochauflösenden 30× Keimbahnanalysen bis hin zu niedrigauflösenden 0,5-2× somatischen Screenings – mit passenden bioinformatischen Pipelines, die für jeden Ansatz optimiert sind.
Was ist Copy Number Variation und warum ist es wichtig?
Kopienzahlvarianten sind strukturelle Veränderungen, bei denen ein DNA-Segment in mehr oder weniger Kopien als im Referenzgenom vorhanden ist. Sie reichen von etwa 1 kb bis zu mehreren Megabasen, und ihre Entstehung wird hauptsächlich durch nicht-allelische homologe Rekombination (NAHR) zwischen flankierenden segmentalen Duplikationen während der Meiose und durch nicht-homologe Endverbindung (NHEJ) sowohl in meiotischen als auch in mitotischen Kontexten vorangetrieben. CNVs werden in Deletionen (Verlust eines genomischen Segments, wodurch die Kopienzahl auf 1 oder 0 reduziert wird) und Duplikationen (Gewinn einer zusätzlichen Kopie, wodurch die Kopienzahl auf 3 oder mehr erhöht wird) klassifiziert. Diese Klassifikation ist anhand von Sequenzierungsdaten durch die Größe und Richtung der Änderung der Lesetiefenänderung nachweisbar – eine heterozygote Deletion reduziert die erwartete Tiefe um 50 %, während eine homozygote Deletion sie auf null reduziert.
Die biologischen Auswirkungen von CNVs sind erheblich. In der Keimbahn sind CNVs eine gut etablierte Ursache genetischer Störungen: Das 22q11.2-Mikrodeletionssyndrom (1 von 4.000 Lebendgeburten), die Charcot-Marie-Tooth-Krankheit (PMP22-Duplikation) und das Smith-Magenis-Syndrom (RAI1-Deletion) sind klassische Beispiele. Studien im Bevölkerungsmaßstab schätzen, dass große CNVs (>50 kb) etwa 15 % des Genoms in der Kopienzahl betreffen und mehr interindividuelle genetische Variation verursachen als alle SNVs zusammen. Bei Krebs treiben fokale Amplifikationen von Onkogenen wie MYC, EGFR, KRAS und ERBB2 direkt das Tumorwachstum voran, während homozygote oder heterozygote Deletionen von Tumorsuppressorgenen wie TP53, CDKN2A, PTEN und RB1 kritische regulatorische Wege eliminieren. Variant-Calling-Dienste Fügen Sie die CNV-Erkennung als Standardkomponente der umfassenden genomischen Analyse hinzu.
Wie die CNV-Erkennung aus Sequenzierungsdaten funktioniert — Die algorithmische Grundlage
Alle sequenzierungsbasierten CNV-Erkennungsmethoden teilen ein gemeinsames algorithmisches Rahmenwerk, das auf der Analyse der Lesetiefe basiert, obwohl die spezifischen Implementierungen zwischen den Werkzeugen erheblich variieren. Das Verständnis dieses Rahmens ist entscheidend für die Interpretation von CNV-Ergebnissen und die Fehlersuche bei fehlgeschlagenen Analysen.
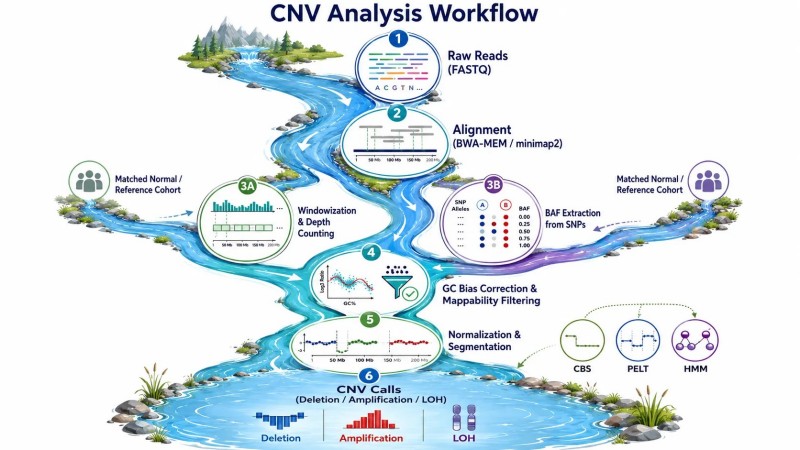
Tiefezählung und FensterungSequenzierte Reads werden an das Referenzgenom ausgerichtet, und die Anzahl der Reads, die auf jedes genomische Fenster abgebildet werden, wird gezählt. Die Fenstergröße ist ein kritischer Parameter – kleinere Fenster (100 bp bis 1 kb) bieten eine höhere Auflösung für Bruchpunkte, aber eine geringere statistische Power pro Fenster, während größere Fenster (10-100 kb) das Signal-Rausch-Verhältnis erhöhen, jedoch auf Kosten verschwommener Bruchpunkte. Für WGS bei 30× bieten 1 kb Fenster ausreichend Power für die CNV-Erkennung. Für LP-WGS bei 1× sind Fenster von 100-500 kb erforderlich. Die Read-Tiefe in jedem Fenster folgt einer Poisson-Verteilung mit einem Mittelwert, der der erwarteten Abdeckung entspricht, und CNVs werden als Regionen identifiziert, in denen die beobachtete Tiefe nach der Normalisierung signifikant von dieser Erwartung abweicht.
Normalisierung — Beseitigung technischer VariationenDie rohe Lesetiefe wird von technischen Faktoren dominiert, die nicht mit der Kopienzahl zusammenhängen. Der GC-Gehalt allein führt zu einer 2- bis 5-fachen Spannbreite in der Abdeckungstiefe – GC-reiche Regionen sequenzieren in den meisten Bibliotheksvorbereitungsprotokollen mit höherer Effizienz, was systematische Spitzen und Täler erzeugt, die in Proben aus demselben Sequenzierungslauf konsistent sind. Die Normalisierung korrigiert dies, indem sie das Verhältnis von beobachteter zu erwarteter Tiefe für Fenster mit ähnlichem GC-Gehalt berechnet und dann die GC-Bias-Kurve glättet, um feinkörniges Rauschen zu entfernen. Die Mappability-Korrektur schließt Fenster aus, in denen ein erheblicher Teil des Genoms nicht eindeutig durch kurze Reads kartiert werden kann – typischerweise Zentromere, Telomere und segmentale Duplikationen. Ohne diese Korrekturen würde das GC-Bias-Signal falsche CNV-Anrufe in den meisten GC-reichen und GC-arme Regionen des Genoms erzeugen.
SegmentierungNach der Normalisierung wird das Tiefenprofil mithilfe von Segmentierungsalgorithmen in Segmente mit konsistenter Kopienzahl unterteilt. Die zirkuläre binäre Segmentierung (CBS), die im DNAcopy R-Paket implementiert ist, teilt das Genom rekursiv in Segmente, indem benachbarte Fenster auf signifikante Unterschiede in der mittleren Tiefe getestet werden. Der PELT-Algorithmus (Pruned Exact Linear Time), der von GATK gCNV verwendet wird, ist schneller und skaliert linear mit der Anzahl der Fenster. Verborgene Markov-Modelle (HMMs), die von XHMM für Exomdaten verwendet werden, behandeln den Kopienzahlzustand als verborgene Variable, die aus der beobachteten Tiefenfolge abgeleitet wird. Die Wahl des Segmentierungsalgorithmus beeinflusst das Gleichgewicht zwischen Sensitivität (Erkennung kleiner CNVs) und Spezifität (Vermeidung der Übersegmentierung des Genoms in viele kleine Segmente, die eher Rauschen als echte CNVs widerspiegeln). In der Praxis liefert CBS konservativere Ergebnisse mit weniger falsch positiven Ergebnissen, während HMMs empfindlicher sind, jedoch auf Kosten einer erhöhten Rate falsch positiver Ergebnisse bei Einzel-Fenster-Ereignissen.
Genotypzuweisung aus der B-Allel-HäufigkeitNeben der Lesetiefe liefern heterozygote SNP-Positionen in den ausgerichteten Reads Informationen zur B-Allel-Häufigkeit (BAF). In diploiden Regionen gruppiert sich die BAF bei 0,5 für heterozygote SNPs. In Regionen mit Änderungen der Kopienzahl weicht die BAF von 0,5 ab – der Verlust der Heterozygotie (LOH) verschiebt die BAF in Richtung 0 oder 1, während die Amplifikation sie in einem Muster verschiebt, das vom Allel-Kopienverhältnis abhängt. Werkzeuge wie Control-FREEC und Canvas integrieren BAF zusammen mit der Tiefe, um CNV-Typen (kopieneutrale LOH vs. echte Deletion) zu unterscheiden und CNVs in Proben mit normaler Zellkontamination zu erkennen, wo das Signalsignal allein mehrdeutig sein kann.
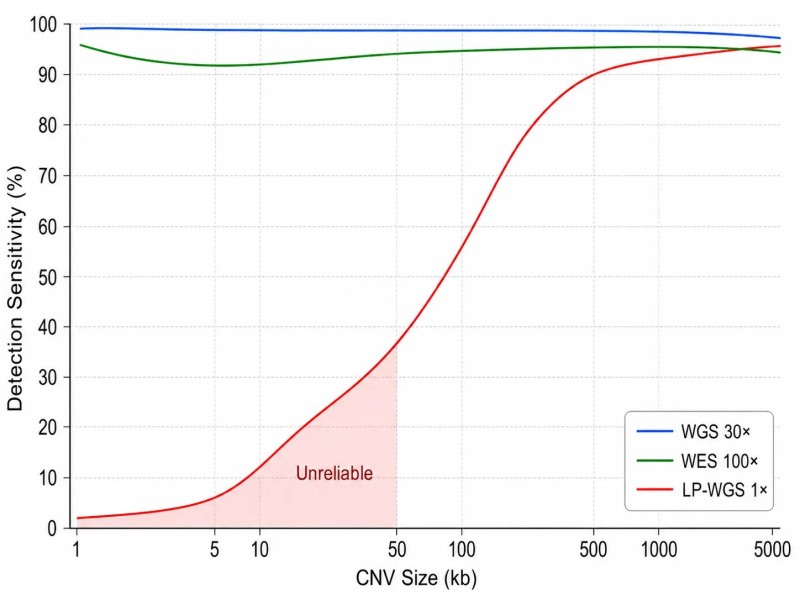
Die quantitative Beziehung zwischen Tiefe und CNV-SignalstärkeDie Zuverlässigkeit eines CNV-Calls hängt vom Signal-Rausch-Verhältnis der beobachteten Tiefenabweichung ab. Bei einer heterozygoten Deletion beträgt die erwartete Tiefenreduktion 50 %. Die Standardabweichung der Lesetiefe in einem Fenster beträgt ungefähr sqrt(mean_depth) für Poisson-verteilte Zählungen. Bei 30× WGS mit 1 kb-Fenstern, die ungefähr 30 Reads enthalten, beträgt die erwartete Standardabweichung etwa 5,5 Reads (18 %), was eine 50%ige Reduktion auf etwa 9 Standardabweichungen vom Mittelwert bringt — leicht nachweisbar. Bei 1× LP-WGS mit 200 kb-Fenstern, die ~200 Reads enthalten, beträgt die erwartete Standardabweichung etwa 14 Reads (7 %), und dasselbe CNV erzeugt eine 50%ige Reduktion, die bei ~7 Standardabweichungen nachweisbar ist. Nachdem jedoch die GC-Korrektur und Normalisierung die systematischen Verzerrungen beseitigt haben, ist das verbleibende Rauschen in LP-WGS ungefähr 2-3× höher als die Poisson-Erwartung aufgrund von Fragmentierungsvariabilität und Alignierungsartefakten. Dieses zusätzliche Rauschen ist der Grund, warum LP-WGS größere Fenster als WGS für eine äquivalente Nachweisempfindlichkeit benötigt. Das Verständnis dieser quantitativen Beziehung hilft Forschern, realistische Erwartungen an die CNV-Erkennung zu setzen — es gibt einen direkten Kompromiss zwischen CNV-Größe, Sequenzierungstiefe und Nachweiszuverlässigkeit, der nicht allein durch verbesserte bioinformatische Normalisierung überwunden werden kann.
Abbildung 1: Vier Ansätze zur CNV-Erkennung — Genomabdeckung, Auflösung und optimale Tiefe

Sequenzierungsbasierte CNV-Erkennung – Vier Ansätze im Vergleich
Die CNV-Erkennung aus Sequenzierungsdaten kann mit vier Ansätzen durchgeführt werden, die sich grundlegend in Bezug auf Genomabdeckung, Tiefe und Kosten unterscheiden. Die Wahl zwischen ihnen bestimmt, welche Arten und Größen von CNVs erkannt werden können.
WGS bei 30× AbdeckungDie Ganzgenomsequenzierung in Standardtiefe bietet die umfassendste CNV-Erkennung. Die Lesetiefe wird über das gesamte Genom in Fenstern von 100 bp bis 1 kb gemessen, korrigiert für GC-Gehalt und Kartierbarkeit, und segmentiert, um Regionen mit signifikant verschobener Abdeckung zu identifizieren. WGS bei 30× erkennt heterozygote Deletionen von nur 1-5 kb und Amplifikationen von nur 5-10 kb und deckt sowohl kodierende als auch nicht-kodierende Regionen ab. Der Nachteil sind die Sequenzierungskosten – etwa 90-100 Gb pro Genom – was den Probendurchsatz für große Kohortenstudien einschränkt. Innerhalb des kodierenden Anteils des Genoms beträgt die Auflösung typischerweise 1-2 kb, was ausreicht, um CNVs in einzelnen Exonen zu erkennen.
WES bei 100-200×Die Ganzexom-Sequenzierung erfasst nur den kodierenden Anteil des Genoms (~1-2%, ungefähr 35 Mb), aber die höhere Lesetiefe bietet eine bessere statistische Power zur CNV-Erkennung innerhalb der erfassten Regionen zu vergleichbaren Kosten im Vergleich zur WGS. Die grundlegende Herausforderung der WES-basierten CNV-Erkennung ist die nicht uniforme Abdeckung, die dem Target-Capture innewohnt – die Hybridisierungseffizienz variiert zwischen den Sonden und zwischen den Regionen innerhalb desselben Sonden-Sets, was systematisches Rauschen einführt, das spezifisch für die Probe ist und nicht vollständig durch generische GC-Normalisierung korrigiert werden kann. ECOLE (2023, Nature Communications), ein auf Deep Learning basierender CNV-Caller für WES-Daten, geht dies an, indem ein konvolutionales neuronales Netzwerk auf simulierten Daten trainiert wird, die das capture-spezifische Rauschprofil jedes Kits berücksichtigen, und erzielt 20-30% weniger falsch-positive Ergebnisse als herkömmliche WES-CNV-Caller. Für Forscher, die WES zur CNV-Analyse verwenden, wird eine minimale mittlere Zielabdeckung von 100× empfohlen, wobei mindestens 30 normale Proben in das Projekt zur Referenzkonstruktion einbezogen werden sollten. Whole-Exom-Sequenzierungsdienste Bieten Sie eine Abdeckung von 150-200× für ein CNV-optimiertes WES-Studiendesign an.
LP-WGS bei 0,5-5×Low-Pass-Ganzgenomsequenzierung (LP-WGS) sequenziert das gesamte Genom mit einem Bruchteil der standardmäßigen Tiefe und ist damit die kosteneffektivste Methode zum Screening von CNVs. Bei einer Abdeckung von 1×, etwa 3 Gb pro Probe, erkennt LP-WGS CNVs, die größer als 50-100 kb sind, mit einer Sensitivität, die mit der chromosomalen Mikroarray vergleichbar ist – was es zu einer praktikablen Alternative für klinisches CNV-Screening macht, wo die Auflösungsanforderungen moderat sind. Ein Benchmark von 2025 in Briefings in Bioinformatics zeigte, dass LP-WGS bei 1× mit 200 kb Fenstern eine Sensitivität von über 90 % für Deletionen >100 kb und über 85 % für Duplikationen >150 kb erreicht. Der Parameter der Fenstergröße ist der entscheidende Hebel – größere Fenster verbessern die Sensitivität auf Kosten der Auflösungsfähigkeit der Bruchpunkte, und die optimale Einstellung skaliert umgekehrt mit der Tiefe (200 kb bei 1×, 50 kb bei 5×). Für Projekte, die CNV-Erkennung mit der Probendurchsatzrate in Einklang bringen müssen, bietet LP-WGS die besten Kosten pro Probe.
Langzeit-Sequenzierung (PacBio HiFi / Nanopore)Lange Reads, die 10-20 kb (HiFi) oder über 100 kb (Nanopore) umfassen, nähern sich der CNV-Erkennung aus einem grundsätzlich anderen Prinzip: Anstatt die Kopienzahl aus der Tiefe abzuleiten, können sie physisch die CNV-Bruchpunkte überbrücken und bieten eine Basis-Paar-Auflösung der Bruchstellenverbindung. Dies ist besonders wertvoll für CNVs in repetitiven Regionen—segmentale Duplikationen, die MHC-Region, tandemartige Genanordnungen—wo die Signale der Short-Read-Tiefe unzuverlässig sind. Der HiFiCNV-Caller von PacBio (2024) ist das erste Tool, das für die CNV-Erkennung mit langen Reads optimiert ist. Ein Benchmark aus dem Jahr 2024 ergab, dass HiFi-Reads bei 15× etwa 30 % mehr CNVs erkannten als Short-Read-WGS bei 30× in denselben Proben, wobei zusätzliche Aufrufe sich auf segmentale Duplikationen konzentrierten. Der Nachteil sind die Kosten—die Sequenzierung mit langen Reads bei vergleichbarer Genomabdeckung ist 3-5× teurer als Methoden mit kurzen Reads.
Abbildung 2: Sensitivität der CNV-Erkennung in Abhängigkeit von der Sequenzierungstiefe und der CNV-Größe
Bioinformatik-Tools zur CNV-Erkennung — Algorithmische Grundlagen und Benchmark-Leistung
Eine Benchmark-Studie aus dem Jahr 2024 in Genome Biology bewertete sechs CNV-Calling-Tools an einer hyper-diploiden Krebszelllinie (HCC1395) mit passenden WGS- und WES-Daten und erzeugte umsetzbare Leistungsdaten für die Auswahl der Tools.
CNVkitEntwickelt für WES-Daten mit gepaarten Tumor-Normal-Proben. CNVkit erstellt ein gepooltes Referenzset aus Normalproben, korrigiert für GC-Bias und segmentiert das Abdeckungsignal mithilfe der zirkulären binären Segmentierung. Es ist das am weitesten validierte WES-CNV-Tool in der Krebsgenomik. Benchmark-Ergebnis: >90% Präzision bei der Erkennung somatischer CNVs in WES mit passenden Normalproben. Am besten geeignet für: projektbezogene Krebs-WES-Analysen mit >10 Normalproben.
GATK gCNVEntwickelt für die Erkennung von Keimbahn-CNVs (Copy Number Variations) bei der WGS (Whole Genome Sequencing) auf Bevölkerungsebene. Verwendet ein Bayessches Modell mit PELT-Segmentierung, das das Abdeckungsprofil aus einer Kohorte lernt (ohne dass für jede Probe passende normale Kontrollen erforderlich sind). Standard für große WGS-Kohorten (über 100 Proben). Benchmark-Ergebnis: höchste Rückrufquote (>85%) für seltene Keimbahn-CNVs aufgrund kohortenbewusster Rauschunterdrückung. Am besten geeignet für großangelegte WGS-Studien zu Keimbahn-CNVs, bei denen keine normalen Kontrollen verfügbar sind.
Control-FREECIdentifiziert CNVs sowohl anhand der Lese-Tiefe als auch der B-Allel-Häufigkeit, was die Erkennung in Abwesenheit von passenden Normal-Kontrollen ermöglicht. Die allelspezifische Fähigkeit ist nützlich für Proben mit normaler Zellkontamination – BAF kann CNVs aufdecken, wo die Tiefe allein mehrdeutig ist. Benchmark-Ergebnis: moderate Präzision (~80%), aber beste Flexibilität über Datentypen hinweg. Am besten geeignet für: Projekte mit begrenzten Kontrollproben oder wo allelische Informationen benötigt werden.
LeinwandIlluminas empfohlener CNV-Caller für WGS und WES. Integriert die Tiefe mit GC-/Mappability-Korrektur und BAF aus SNP-Reads. Optimiert für die Illumina DRAGEN-Pipeline. Benchmark-Ergebnis: >90 % Präzision für somatische CNVs bei WGS, vergleichbar mit CNVkit für WES. Am besten geeignet für: Nur-Illumina-Workflows und automatisierte Pipeline-Integration.
SCHULE: Auf Deep Learning basierender WES CNV-Caller (konvolutionales neuronales Netzwerk). Trainiert mit simulierten Daten mit erfassungsspezifischem Rauschen, erreicht niedrigere falsch-positive Raten als CNVkit für Single-Exon-CNVs. Benchmark-Ergebnis: 20-30% weniger falsch-positive Ergebnisse als CNVkit für WES. Am besten geeignet für: WES-Projekte, die hohe Spezifität erfordern, insbesondere wenn die Validierungskapazität begrenzt ist.
HiFiCNVPacBio's Long-Read-CNV-Caller für HiFi-Lesungen. Segmentiert die Abdeckung von Langlesungen nach GC-Korrektur und profitiert von der hohen Kartierbarkeit von Langlesungen. Werkzeug in der frühen Entwicklungsphase. Benchmark-Ergebnis: erkennt ~30% mehr CNVs in repetitiven Regionen als Methoden mit Kurzlesungen. Am besten geeignet für: Projekte, die PacBio HiFi-Daten verwenden und eine CNV-Erkennung in komplexen genomischen Regionen erfordern.
CNV-Erkennungsdienste Unterstützen Sie jedes dieser Werkzeuge mit validierten Arbeitsabläufen, die es Forschern ermöglichen, das geeignete Werkzeug basierend auf ihrem Datentyp und ihren Projektzielen auszuwählen.
Abbildung 3: CNV-Bioinformatik-Pipeline — wichtige Verarbeitungsschritte von Rohdaten zu Kopienzahlaufrufen
Herausforderungen, die die Genauigkeit der CNV-Erkennung beeinträchtigen
Die CNV-Erkennung ist empfindlicher gegenüber technischen Artefakten als die SNV-Erkennung, da die Lese-Tiefe von mehreren Faktoren unabhängig von der biologischen Kopienzahl beeinflusst wird. Das Verständnis dieser Störfaktoren und die Anwendung geeigneter Korrekturen sind entscheidend, um zuverlässige Ergebnisse zu erzielen.
GC-BiasDie Effizienz der PCR-Amplifikation variiert mit dem GC-Gehalt über einen Bereich von 2- bis 5-fach, was systematische Tiefenvariationen erzeugt, die CNV-Signale nachahmen. Die GC-Korrektur berechnet die beobachteten zu erwartenden Tiefenverhältnisse innerhalb von GC-abgestimmten Fenstern, jedoch ist diese Korrektur für fragmentierte oder DNA-Proben mit niedrigem Input unvollkommen. Der verbleibende Bias nach der Korrektur macht einen erheblichen Anteil der falsch-positiven CNV-Anrufe in sowohl WGS- als auch WES-Daten aus, insbesondere in GC-reichen Promotorregionen und GC-armen intergenen Regionen.
KartierbarkeitEtwa 10-15% des menschlichen Genoms—Zentromere, Telomere, ribosomale DNA-Arrays und segmentale Duplikationen—können mit kurzen Reads nicht eindeutig kartiert werden und müssen von der CNV-Analyse ausgeschlossen werden. CNVs in diesen Regionen werden systematisch übersehen. Bei WES hängt der unzugängliche Anteil vom Design des Capture-Kits ab und kann 15-20% der anvisierten Regionen erreichen.
Entspricht den normalen AnforderungenDie Identifizierung von CNVs aus der Tiefe eines einzelnen Samples ohne Referenzvergleich – Single-Sample-CNV-Calling – hat eine begrenzte Genauigkeit, da technische Tiefenvariationen nicht von biologischen CNV-Signalen isoliert unterschieden werden können. Standardmäßig wird entweder eine passende normale Kontrolle (somatische Krebsfälle) oder ein gepooltes Referenzset aus ≥10 normalen Proben (Keimbahn-WGS) oder ≥30 Proben (Keimbahn-WES) verwendet. Projekte, die diese Mindestanforderungen erfüllen, verwenden Werkzeuge mit integrierter modellbasierter Normalisierung (GATK gCNV) als Alternative.
FFPE-ArtefakteFFPE-DNA hat durchschnittliche Fragmentgrößen von <300 bp und deaminierte Basen aufgrund von Formalinvernetzung. Diese Eigenschaften erhöhen die Tiefenvarianz und reduzieren das Signal-Rausch-Verhältnis bei CNV. Ein Benchmark aus 2024 ergab, dass die Präzision der CNV-Erkennung bei FFPE im Vergleich zu frisch gefrorenem Gewebe um 15-25% abnimmt. Milderungsstrategien umfassen passende FFPE-Normal-Kontrollen, erhöhte Sequenzierungstiefe und spezialisierte Normalisierungsmethoden für fragmentierte DNA.
Tumorreinheit und HeterogenitätIn Krebsproben ist das effektive CNV-Signal das Produkt aus dem CNV-Zustand, dem Anteil der Tumorzellen, die dieses CNV tragen, und der Probenreinheit. Ein CNV, das in 40% der Tumorzellen einer 60% reinen Probe vorhanden ist, erzeugt eine Tiefenänderung von nur 12% vom diploiden Zustand – ununterscheidbar von Rauschen in den meisten Pipelines. BAF-basierte Werkzeuge (Control-FREEC) ermöglichen die Erkennung bei einer Reinheit von bis zu ~20%, während Methoden, die nur auf der Tiefe basieren, >30% für die somatische CNV-Erkennung benötigen.
Niedrigabdeckungsgrenzen für spezifische CNV-TypenVerschiedene CNV-Typen haben bei gleicher Tiefe unterschiedliche Nachweisgrenzen. Amplifikationen (Zunahmen) erzeugen schwächere Signale als Deletionen – eine Triplikation führt nur zu einer 50%igen Erhöhung der erwarteten Tiefe im Vergleich zu dem 50%igen Rückgang einer Deletion mit einer Kopie. Bei 30× WGS sind beide nachweisbar, aber bei 1× LP-WGS erfordern Amplifikationen 2-3× größere Ereignisse, um vergleichbare Sicherheit zu erreichen. Homozygote Deletionen erzeugen das stärkste Signal (Tiefe, die am Ziel gegen null geht) und sind selbst bei den niedrigsten Tiefen nachweisbar. Das Verständnis dieser typenspezifischen Nachweisgrenzen ist wichtig für die Planung von Projekten, die darauf abzielen, spezifische Klassen von CNVs zu erkennen.
Abbildung 4: Herausforderungen bei der CNV-Analyse von Tumoren – die kombinierte Wirkung von Reinheit, Probenqualität und Sequenzierungstiefe

CNV-Analyse bei Krebs — Somatische vs. Keimbahnüberlegungen
Die somatische CNV-Erkennung bei Krebs unterscheidet sich in drei wesentlichen Punkten von der Erkennung von Keimbahn-CNVs: der Notwendigkeit einer passenden Normalprobe, dem Vorhandensein subklonaler CNVs und dem verwirrenden Effekt von Tumorploidie-Änderungen.
Eine passende normale Probe (Blut oder angrenzendes normales Gewebe) ist entscheidend, um somatische CNVs von vererbten Keimbahn-CNVs und technischen Artefakten zu unterscheiden. Die passende Normale liefert stichprobenbezogene GC-Bias, Erfassungs-effizienz und Korrektur der Sequenzierungs-Lauf-Effekte. In ihrer Abwesenheit können gepoolte normale Referenzen teilweise kompensieren, jedoch mit reduzierter Sensitivität. Für Krebsstudien wird LP-WGS bei 1-2× schnell für die somatische CNV-Profilierung in großen Kohorten angenommen – eine Studie von 2025 mit 2.000 Krebsproben ergab, dass LP-WGS bei 1× fokale Amplifikationen und homozygote Deletionen mit >85% Übereinstimmung mit 30× WGS für Ereignisse größer als 100 kb nachwies.
Kopienzahlensignaturen – genomweite Muster von Amplifikationen und Deletionen, die mit spezifischen mutationalen Prozessen verknüpft sind – liefern Informationen über individuelle Genveränderungen hinaus. Der HRD (Defizienz der homologen Rekombination) Score, der aus genomweiten CNV-Mustern abgeleitet wird, hat sich als prädiktiver Biomarker für die Reaktion auf PARP-Inhibitor-Therapien etabliert. Klinische WGS-Dienste Unterstützung sowohl für Standard- als auch für Niedrigpasskonfigurationen zur Analyse von Krebs-CNV mit verfügbaren gepaarten Protokollen für Tumor und Normalgewebe.
Einzelzell-CNV-ErkennungEine aufkommende Grenze in der CNV-Analyse ist das Single-Cell-CNV-Profiling, das die intra-tumorale Heterogenität auflöst, die in der Bulk-Sequenzierung gemittelt wird. Single-Cell-WGS mit niedriger Tiefe (0,1-1× pro Zelle) in Kombination mit Algorithmen zur Kopienzahl-Inferenz kann die klonale Architektur eines Tumors rekonstruieren, indem CNV-Unterschiede zwischen einzelnen Zellen erkannt werden. Dieser Ansatz wurde verwendet, um metastatische Verbreitungsmuster nachzuvollziehen und seltene Subklone zu identifizieren, die resistenzverleihende CNVs tragen, die in der Bulk-Analyse unsichtbar sind. Der Nachteil ist, dass Einzelzell-Daten pro Zelle mehr Rauschen aufweisen als Bulk-Daten, was spezialisierte Normalisierungs- und Anrufalgorithmen sowie eine größere Zellanzahl für eine zuverlässige CNV-Zuordnung erfordert. Projekte, die eine Single-Cell-CNV-Analyse in Betracht ziehen, sollten mindestens 100-500 Zellen pro Probe einplanen, um eine ausreichende statistische Power für die Subklon-Erkennung zu erreichen.
Abbildung 5: Auswahlleitfaden für CNV-Analysewerkzeuge — Anpassung des Werkzeugs an Datentyp und Forschungsziel

Langzeit-Sequenzierung zur CNV-Erkennung
Long-Read-Sequenzierung behebt die grundlegende Einschränkung der CNV-Erkennung mit Kurzlesungen: die Unfähigkeit, Lesevorgänge auf sich wiederholende Regionen abzubilden und Breakpoints zu überbrücken. PacBio HiFi-Lesungen mit 10-20 kb und einer Genauigkeit von über 99,9 % können direkt CNV-Breakpoints überbrücken, während Nanopore-Ultra-Long-Lesungen von über 100 kb gesamte komplexe Umstellungen überbrücken können.
Ein Benchmark von 2024 zur Erkennung von CNVs mit langen Reads hat ergeben, dass HiFi-Reads bei 15× etwa 30 % mehr CNVs in denselben Proben erkannten als Short-Read-WGS bei 30×, wobei zusätzliche Aufrufe in segmentalen Duplikationen und anderen repetitiven Regionen konzentriert waren. Die Auflösung der Bruchpunkte verbesserte sich von 1-10 kb (Short-Read) auf innerhalb von 100 bp (HiFi).
Die Detektion von langen CNVs ist besonders wertvoll, wenn der Forschungsschwerpunkt komplexe genomische Regionen umfasst—segmentale Duplikationen, die MHC-Region, tandemduplizierte Genfamilien oder bekannte CNV-Hotspots. Für Projekte, die sich auf die >90 % des Genoms konzentrieren, die mit kurzen Reads zugänglich sind, bleiben Methoden mit kurzen Reads aufgrund der geringeren Kosten und der ausgereifteren Werkzeuge die praktische Wahl.
Hybride Strategien für eine umfassende CNV-AnalyseFür Projekte, die sowohl kosteneffektives genomweites Screening als auch hochauflösende Breakpoint-Analysen erfordern, bietet ein hybrider Ansatz, der kurzes LP-WGS bei 1-2× für die anfängliche CNV-Identifizierung mit gezielter Langzeit-Sequenzierung von CNV-Breakpoint-Regionen kombiniert, die effizienteste Ressourcennutzung. Das LP-WGS-Screening identifiziert Kandidaten-CNVs und schätzt deren ungefähre Grenzen, während die Langzeit-Sequenzierung den genauen Breakpoint auflöst und die zugrunde liegende Sequenzarchitektur identifiziert (z. B. NAHR zwischen spezifischen Wiederholungselementen). Dieser gestufte Ansatz wurde erfolgreich in klinischen CNV-Validierungs-Pipelines und in Bevölkerungsstudien eingesetzt, in denen eine umfassende CNV-Charakterisierung erforderlich ist, aber Kosteneinschränkungen die Verwendung von genomweitem Langzeit-Sequenzieren begrenzen.
CNV-Validierung — Die Rolle orthogonaler Methoden
Computational CNV-Calls sollten durch orthogonale Methoden validiert werden, bevor starke biologische Schlussfolgerungen gezogen werden. Digitale Tropfen-PCR (ddPCR) bietet eine absolute Quantifizierung der Kopienzahl an spezifischen Loci und ist praktisch für die Validierung von 5-20 Kandidaten pro Projekt, mit einer Nachweisempfindlichkeit, die ausreicht, um Einzelkopiengewinne oder -verluste in Proben mit 50 % oder höherer Tumorpurity zu bestätigen. Chromosomale Mikroarray-Analyse (aCGH) bleibt der goldene Standard im gesamten Genom mit >95 % Sensitivität und Spezifität für CNVs >50 kb und dient als Referenzplattform für die meisten klinischen CNV-Validierungs-Pipelines. Für Projekte, die neuartige krankheitsassoziierte CNVs oder klinische Ergebnisse berichten, ist die Validierung durch mindestens eine orthogonale Methode gängige Praxis vor der Veröffentlichung oder klinischen Berichterstattung.
CNV-Datenbanken für Annotation und Interpretation
UCSC-GenombrowserPrimäre Visualisierungsplattform für CNV-Aufrufe im genomischen Kontext, mit Spuren für Wiederholungselemente, bekannte Gene, segmentale Duplikationen und die CNV-Häufigkeit in der Bevölkerung aus 1000 Genomes und gnomAD.
DECIPHER-DatenbankKuratiert CNVs, die mit genetischen Störungen assoziiert sind, und verknüpft jede Variante mit klinischen Phänotypen und Geninhalten zur Beurteilung der Pathogenität.
ClinGen-DatenbankGenebasierte Dosissensitivitätsscores – systematische Bewertungen von Haploinsuffizienz und Triplosensitivität für jedes Gen – unterstützen die Interpretation, ob ein genhaltiger CNV wahrscheinlich pathogen ist. Genomdatenanalyse-Dienste integrieren Sie diese Datenbanken in automatisierte Annotationsworkflows.
Rechenressourcen für CNV-Analyse
Die CNV-Analyse ist im Vergleich zur WGS-Ausrichtung oder de novo Assemblierung rechnerisch bescheiden. Eine 30× WGS-Probe benötigt nach der Ausrichtung etwa 1-2 Stunden für die CNV-Erkennung auf einem Standard-Compute-Knoten. Die CNV-Erkennung bei WES mit CNVkit erfordert 30-60 Minuten pro Probe. LP-WGS bei 1× verarbeitet in 15-30 Minuten. GATK gCNV erfordert eine Verarbeitung auf Kohortenebene — nachdem das Modell aus der Kohorte erstellt wurde (1-2 Stunden), erfolgt die individuelle Probenanrufung schnell (10-20 Minuten pro Probe). Die Speicheranforderungen werden von den ausgerichteten BAM/CRAM-Dateien dominiert, die für die Tiefenextraktion benötigt werden. Projekte, die Cloud-Computing nutzen, sollten die Rechenressourcen basierend auf der Anzahl der Proben und dem Datenvolumen budgetieren.
Abbildung 6: CNV-Analyse-Projektfahrplan — von der Forschungsfrage zur CNV-Interpretation
Häufig gestellte Fragen
Welche Sequenzierungstiefe ist für die CNV-Erkennung aus WGS erforderlich?
Zur Erkennung von CNVs, die größer als 5-10 kb im gesamten Genom sind, sind 15-30× Abdeckung Standard. LP-WGS mit 1-2× erkennt CNVs, die größer als 50-100 kb sind, für klinische Screenings, bei denen Kosteneffizienz priorisiert wird.
Wie beeinflusst die Qualität von FFPE-Proben die CNV-Erkennung?
FFPE-Proben haben fragmentierte DNA und Basisschäden, die das Rauschlevel der Lesetiefe erhöhen und die Sensitivität zur Erkennung von CNVs um 15-25 % im Vergleich zu frisch gefrorenem Gewebe verringern. Die Verwendung von passenden FFPE-Normalproben und die Erhöhung der Sequenzierungstiefe kompensieren dies teilweise.
Was ist die minimale Tumorpurität für die Erkennung somatischer CNVs?
Die meisten Werkzeuge erfordern eine Reinheit von über 20-30% für eine zuverlässige Erkennung. BAF-basierte Werkzeuge (Control-FREEC) schneiden bei niedriger Reinheit besser ab als Methoden, die nur auf der Tiefe basieren.
Wie wähle ich zwischen CNVkit und GATK gCNV?
GATK gCNV wird für die großangelegte WGS-Germline-CNV-Erkennung empfohlen, bei der ein Populationsmodell erstellt werden kann. CNVkit wird für projektbezogene WES-Krebs-CNV mit passenden Normal-Kontrollen empfohlen.
Kann ich CNVs aus RNA-Seq-Daten erkennen?
Die CNV-Erkennung durch RNA-Seq ist möglich, aber weniger zuverlässig als DNA-basierte Methoden aufgrund von Ausdrucksvariationen. Eine Validierung durch DNA-basierte Methoden wird für alle RNA-Seq-CNV-Funde empfohlen.
Was ist die minimale Referenzgröße für die Analyse von gepoolten normalen CNVs?
Mindestens 10 normale Proben für WGS und 30 für WES, um das höhere Rauschlevel der Zielerfassung widerzuspiegeln. Unterhalb dieser Schwellenwerte werden modellbasierte Methoden (GATK gCNV) bevorzugt.
Referenzen
- Bewertung der Erkennung somatischer Kopienzahlvariationen durch NGS-Technologien und bioinformatische Werkzeuge in einem hyperdiploiden Krebsgenom. Genome Biology. 2024;25:164.
- ECOLE: Lernen, Kopienzahlvarianten aus Daten der gesamten Exomsequenzierung zu identifizieren. Nature Communications. 2023;14:44116.
- Benchmarking der Erkennung von Kopienzahlvariationen mit niedrigdeckender Ganzgenomsequenzierung. Briefings in Bioinformatics. 2025;26:bbaf514.
- Lang und präzise: Wie HiFi-Sequenzierung die genomische Forschung transformiert. Genomik, Proteomik & Bioinformatik. 2025;23:qzaf003.
- Low-Pass-Ganzgenomsequenzierung ist eine zuverlässige und kosteneffiziente Methode zur Erkennung von CNVs. Annals of Human Genetics. 2023;87:251-260.
Verwandte Dienstleistungen
- Whole Genome Sequenzierung
- Whole Exome Sequencing - Gesamtes Exom-Sequenzierung
- Variantenaufruf
- Nächste Generation Sequenzierung
- Genomdatenanalyse
- Bioinformatik-Dienstleistungen
- WGS-Dienste
- WES-Dienste
Nur zu Forschungszwecken, nicht zur klinischen Diagnose, Behandlung oder individuellen Gesundheitsbewertung bestimmt.


